class: center, middle, inverse, title-slide .title[ # Single-cell RNA sequencing ~ Session 2<br /> <html><br /> <br /> <hr color='#EB811B' size=1px width=796px><br /> </html> ] .author[ ### Rockefeller University, Bioinformatics Resource Centre ] .date[ ### <a href="http://rockefelleruniversity.github.io/scRNA-seq/" class="uri">http://rockefelleruniversity.github.io/scRNA-seq/</a> ] --- ## Overview In this course we are going to introduce basic analysis for single-cell RNAseq, with a specific focus on the *10X system*. The course is divided into multiple sessions. In this third session, we will first evaluate methods to remove the effects of ambient RNA as well as detecting doublets within the data. Following this we will work to integrate a dataset with that we processed in session 2 to create a single dataset for further analysis. --- ## The data For these sessions we are going to make use of two datasets. The first set will be from the recent paper [**Enteroendocrine cell lineages that differentially control feeding and gut motility**](https://elifesciences.org/articles/78512). This contains scRNA data from either Neurod1 and Neurog3 expressing enteroendocrine cells. --- ## Read in the filtered matrix from CellRanger The filtered matrix contains droplets considered to be true cell containing droplets. This was performed using the emptyDrops method integrated into CellRanger which we also review ourselves in session 2 using the original implementation in the DropletUtils package. ``` r h5file <- "https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/SRR_NeuroD1/outs/filtered_feature_bc_matrix.h5" local_h5file <- basename(h5file) download.file(h5file, local_h5file) sce.NeuroD1_filtered <- read10xCounts(local_h5file, col.names = TRUE) sce.NeuroD1_filtered ``` ``` ## class: SingleCellExperiment ## dim: 39905 3468 ## metadata(1): Samples ## assays(1): counts ## rownames(39905): Gm26206 Xkr4 ... MT-TrnT MT-TrnP ## rowData names(3): ID Symbol Type ## colnames(3468): AAACCCACACAATGTC-1 AAACCCACACCACTGG-1 ... ## TTTGTTGTCTCTTAAC-1 TTTGTTGTCTGGACCG-1 ## colData names(2): Sample Barcode ## reducedDimNames(0): ## mainExpName: NULL ## altExpNames(0): ``` --- ## Read in the unfiltered matrix from CellRanger We also will read in the unfiltered matrix containing all Droplets including non-cell containing droplets which will most likely contain only ambient RNAs. ``` r h5file <- "https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/SRR_NeuroD1/outs/raw_feature_bc_matrix.h5" local_h5file <- "NeuroD1_raw_feature_bc_matrix.h5" download.file(h5file, local_h5file) sce.NeuroD1_unfiltered <- read10xCounts(local_h5file, col.names = TRUE) sce.NeuroD1_unfiltered ``` ``` ## class: SingleCellExperiment ## dim: 39905 1244317 ## metadata(1): Samples ## assays(1): counts ## rownames(39905): Gm26206 Xkr4 ... MT-TrnT MT-TrnP ## rowData names(3): ID Symbol Type ## colnames(1244317): AAACCCAAGAAACACT-1 AAACCCAAGAAACCAT-1 ... ## TTTGTTGTCTTTGCTA-1 TTTGTTGTCTTTGTCG-1 ## colData names(2): Sample Barcode ## reducedDimNames(0): ## mainExpName: NULL ## altExpNames(0): ``` --- ## Ambient RNA removal (decontX) There are many methods to remove ambient RNA using the background proportion of our dataset (lower portion of our knee plot). First, we will use decontX package to try and remove ambient RNAs. ``` r library(decontX) ``` --- ## DecontX The decontX package is best implemented by providing the filtered and unfiltered datasets, here in singlecellexperiment format. ``` r sce.NeuroD1_filtered <- decontX(sce.NeuroD1_filtered, background = sce.NeuroD1_unfiltered) saveRDS(sce.NeuroD1_filtered, file = "decontx.RDS") sce.NeuroD1_filtered ``` --- ## DecontX The SingleCellExperiemnt object now contains the **decontXcounts assay slot** containing corrected counts ``` ## <2 x 4> sparse DelayedMatrix object of type "double": ## AAACCCACACAATGTC-1 ... AAACCCAGTGTCCGGT-1 ## Gm26206 0 . 0 ## Xkr4 0 . 0 ``` --- ## DecontX As well as **decontX_contamination** and **decontX_cluster** columns in the colData. ``` ## DataFrame with 3468 rows and 4 columns ## Sample Barcode ## <character> <character> ## AAACCCACACAATGTC-1 ~/Downloads/rerunSRR.. AAACCCACACAATGTC-1 ## AAACCCACACCACTGG-1 ~/Downloads/rerunSRR.. AAACCCACACCACTGG-1 ## AAACCCACACGAGAAC-1 ~/Downloads/rerunSRR.. AAACCCACACGAGAAC-1 ## AAACCCAGTGTCCGGT-1 ~/Downloads/rerunSRR.. AAACCCAGTGTCCGGT-1 ## AAACGAAAGCCTCGTG-1 ~/Downloads/rerunSRR.. AAACGAAAGCCTCGTG-1 ## ... ... ... ## TTTGTTGAGCATGCAG-1 ~/Downloads/rerunSRR.. TTTGTTGAGCATGCAG-1 ## TTTGTTGCACATAACC-1 ~/Downloads/rerunSRR.. TTTGTTGCACATAACC-1 ## TTTGTTGGTCTAACTG-1 ~/Downloads/rerunSRR.. TTTGTTGGTCTAACTG-1 ## TTTGTTGTCTCTTAAC-1 ~/Downloads/rerunSRR.. TTTGTTGTCTCTTAAC-1 ## TTTGTTGTCTGGACCG-1 ~/Downloads/rerunSRR.. TTTGTTGTCTGGACCG-1 ## decontX_contamination decontX_clusters ## <numeric> <factor> ## AAACCCACACAATGTC-1 0.1622808 1 ## AAACCCACACCACTGG-1 0.1736627 2 ## AAACCCACACGAGAAC-1 0.0518798 3 ## AAACCCAGTGTCCGGT-1 0.4642820 1 ## AAACGAAAGCCTCGTG-1 0.2798897 2 ## ... ... ... ## TTTGTTGAGCATGCAG-1 0.1381154 2 ## TTTGTTGCACATAACC-1 0.5403299 2 ## TTTGTTGGTCTAACTG-1 0.0518829 3 ## TTTGTTGTCTCTTAAC-1 0.4834494 2 ## TTTGTTGTCTGGACCG-1 0.2025199 2 ``` --- ## DecontX Also we will have an additional reducedDim **decontX_UMAP**. ``` ## DecontX_UMAP_1 DecontX_UMAP_2 ## AAACCCACACAATGTC-1 8.041984 2.747593 ## AAACCCACACCACTGG-1 -3.266628 6.525007 ``` --- ## DecontX As part of its ambient RNA procedure, decontX has generated clusters and a UMAP projection. We can review these projections to see the structure of ambient RNA in our data. <!-- --> --- ## DecontX We can then assess the degree of ambient RNA contamination in each cell using the **decontX_contamination** colData column. <!-- --> --- ## Overlay DecontX We could also overlay our decontX scores onto the data we created in session2. ``` r sce.NeuroD1_filtered_QCed <- readRDS("sce.NeuroD1_filtered_QCed.RDS") sce.NeuroD1_filtered_QCed$decontX_contamination <- sce.NeuroD1_filtered$decontX_contamination[match(colnames(sce.NeuroD1_filtered_QCed), colnames(sce.NeuroD1_filtered), nomatch = 0)] ``` --- ## Overlay DecontX We can now review whether any of our clusters have a high degree of ambient RNAs. ``` r plotColData(sce.NeuroD1_filtered_QCed, x = "label", y = "decontX_contamination") ``` <!-- --> --- ## Overlay DecontX Now we can review the clusters and ambient RNA contamination within a UMAP. ``` r plotUMAP(sce.NeuroD1_filtered_QCed, colour_by = "label") ``` <!-- --> --- ## Overlay DecontX ``` r plotUMAP(sce.NeuroD1_filtered_QCed, colour_by = "decontX_contamination") ``` <!-- --> --- ## CellBender An alternative an popular method for removing artefacts from your singlecell data is CellBender. CellBender uses machine learning to identify features such as ambient RNA and can provide a filtered matrix such as CellRanger for a starting point. We can run CellBender on our samples with defaults using the example below. ``` cellbender remove-background --input ./rerunSRR_NeuroD1/outs/raw_feature_bc_matrix.h5 --output ./rerunSRR_NeuroD1/outs/cellbender_v0.3.2.h5 ``` --- ## CellBender Note CellBender is **much** faster running on a GPU so if we have one available we can use the --cuda flag to speed things up. ``` cellbender remove-background --input ./rerunSRR_NeuroD1/outs/raw_feature_bc_matrix.h5 --output ./rerunSRR_NeuroD1/outs/cellbender_v0.3.2.h5 --cuda ``` --- ## CellBender CellBender outputs a filtered matrix file in H5 format for data import and among other outputs a HTML file summarising important metrics and CSV file of metrics summarised. [HTML Report](https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/SRR_NeuroD1/outs/cellbender_v0.3.2_report.html) [Metrics](https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/SRR_NeuroD1/outs/cellbender_v0.3.2_metrics.csv) --- ## CellBender import We can import the CellBender filtered matrix as before with the CellRanger matrix. ``` r h5file <- "https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/SRR_NeuroD1/outs/cellbender_v0.3.2_filtered.h5" local_h5file <- basename(h5file) download.file(h5file, local_h5file) sce.NeuroD1_CellBender <- read10xCounts(local_h5file, col.names = TRUE, row.names = "symbol") sce.NeuroD1_CellBender$Sample <- "Nd1" class(sce.NeuroD1_CellBender) ``` ``` ## [1] "SingleCellExperiment" ## attr(,"package") ## [1] "SingleCellExperiment" ``` --- ## CellBender import ``` r sce.NeuroD1_CellBender ``` ``` ## class: SingleCellExperiment ## dim: 39905 4877 ## metadata(1): Samples ## assays(1): counts ## rownames(39905): Gm26206 Xkr4 ... MT-TrnT MT-TrnP ## rowData names(3): ID Symbol Type ## colnames(4877): ACGTAGTCACCCAAGC-1 TCATACTCAAAGGCTG-1 ... ## TTGCATTTCTCCTGAC-1 CGAATTGTCTTAGGAC-1 ## colData names(2): Sample Barcode ## reducedDimNames(0): ## mainExpName: NULL ## altExpNames(0): ``` --- ## QC CellBender Now we have an ambient RNA corrected matrix we can reprocess our dataset as before. First we gather the QC. ``` r is.mito <- grepl("^MT", rowData(sce.NeuroD1_CellBender)$Symbol) sce.NeuroD1_CellBender <- addPerCellQCMetrics(sce.NeuroD1_CellBender, subsets = list(Mito = is.mito)) p1 <- plotColData(sce.NeuroD1_CellBender, x = "sum", y = "detected") p2 <- plotColData(sce.NeuroD1_CellBender, x = "sum", y = "subsets_Mito_percent") p3 <- plotColData(sce.NeuroD1_CellBender, x = "detected", y = "subsets_Mito_percent") gridExtra::grid.arrange(p1, p2, p3, ncol = 3) ``` <!-- --> --- ## Filter CellBender ``` r qc.high_lib_size <- colData(sce.NeuroD1_CellBender)$sum > 125000 qc.min_detected <- colData(sce.NeuroD1_CellBender)$detected < 200 qc.mito <- colData(sce.NeuroD1_CellBender)$subsets_Mito_percent > 25 discard <- qc.high_lib_size | qc.mito | qc.min_detected colData(sce.NeuroD1_CellBender) <- cbind(colData(sce.NeuroD1_CellBender), DataFrame(toDiscard = discard)) sce.NeuroD1_CellBender_QCed <- sce.NeuroD1_CellBender[, sce.NeuroD1_CellBender$toDiscard %in% "FALSE"] ``` --- ## Normalise, PCA and projections ``` r set.seed(100) clust.sce.NeuroD1_CellBender_QCed <- quickCluster(sce.NeuroD1_CellBender_QCed) sce.NeuroD1_CellBender_trimmed <- computeSumFactors(sce.NeuroD1_CellBender_QCed, cluster = clust.sce.NeuroD1_CellBender_QCed) sce.NeuroD1_CellBender_QCed <- logNormCounts(sce.NeuroD1_CellBender_QCed) dec.NeuroD1_CellBender_QCed <- modelGeneVar(sce.NeuroD1_CellBender_QCed) top.NeuroD1_CellBender_QCed <- getTopHVGs(dec.NeuroD1_CellBender_QCed, n = 3000) sce.NeuroD1_CellBender_QCed <- fixedPCA(sce.NeuroD1_CellBender_QCed, subset.row = top.NeuroD1_CellBender_QCed) sce.NeuroD1_CellBender_QCed <- runTSNE(sce.NeuroD1_CellBender_QCed, n_dimred = 30) sce.NeuroD1_CellBender_QCed <- runUMAP(sce.NeuroD1_CellBender_QCed, n_dimred = 30) ``` --- ## Cluster CellBender ``` r library(bluster) clust.louvain <- clusterCells(sce.NeuroD1_CellBender_QCed, use.dimred = "PCA", BLUSPARAM = NNGraphParam(cluster.fun = "louvain", cluster.args = list(resolution = 0.8))) clust.default <- clusterCells(sce.NeuroD1_CellBender_QCed, use.dimred = "PCA") colLabels(sce.NeuroD1_CellBender_QCed) <- clust.louvain colData(sce.NeuroD1_CellBender_QCed)$DefaultLabel <- clust.default ``` --- ## Review CellBender ``` r plotUMAP(sce.NeuroD1_CellBender_QCed, colour_by = "label") ``` <!-- --> --- ## Cell annotation to CellBender We can then add the known annotation again. ``` r eec_paper_meta <- read.delim("https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/GSE224223_EEC_metadata.csv", sep = ";") nrd1_cells <- as.data.frame(eec_paper_meta[eec_paper_meta$Library == "Nd1", ]) nrd1_cells$X <- gsub("_2$", "", nrd1_cells$X) sce.NeuroD1_CellBender_QCed$All_Cell_Types <- nrd1_cells$All_Cell_Types[match(colnames(sce.NeuroD1_CellBender_QCed), nrd1_cells$X, nomatch = NA)] ``` --- ## Cell annotation to CellBender And review this annotation in the UMAP ``` r plotUMAP(sce.NeuroD1_CellBender_QCed, colour_by = "All_Cell_Types") ``` <!-- --> --- ## Doublet detection and removal Doublets occur when two cells are within one droplet. This results in a droplet having the average expression of these two cells. We can remove Doublets from our data using multiple methods. Here we will use scDblFinder ``` r library(scDblFinder) sce.NeuroD1_CellBender_QCed <- scDblFinder(sce.NeuroD1_CellBender_QCed, clusters = colLabels(sce.NeuroD1_CellBender_QCed)) ``` ``` ## 10 clusters ``` ``` ## Creating ~1500 artificial doublets... ``` ``` ## Dimensional reduction ``` ``` ## Evaluating kNN... ``` ``` ## Training model... ``` ``` ## iter=0, 60 cells excluded from training. ``` ``` ## iter=1, 74 cells excluded from training. ``` ``` ## iter=2, 77 cells excluded from training. ``` ``` ## Threshold found:0.578 ``` ``` ## 81 (4.9%) doublets called ``` --- ## Doublet detections and removal We can then review to see which clusters contained Doublets. ``` r plotColData(sce.NeuroD1_CellBender_QCed, x = "label", y = "scDblFinder.score") ``` <!-- --> --- ## Doublet detections and removal We can can also assess the relationship between doublets and other metrics. ``` r p1 <- plotColData(sce.NeuroD1_CellBender_QCed, x = "detected", y = "scDblFinder.score") p2 <- plotColData(sce.NeuroD1_CellBender_QCed, x = "sum", y = "scDblFinder.score") gridExtra::grid.arrange(p1, p2, ncol = 2) ``` <!-- --> --- ## Doublet detections and removal And review doublets in our umaps ``` r p1 <- plotUMAP(sce.NeuroD1_CellBender_QCed, colour_by = "label") p2 <- plotUMAP(sce.NeuroD1_CellBender_QCed, colour_by = "scDblFinder.score") gridExtra::grid.arrange(p1, p2, ncol = 2) ``` <!-- --> --- ## Doublet detections and removal We can then filter out doublets from our data. ``` r sce.NeuroD1_CellBender_QCDbled <- sce.NeuroD1_CellBender_QCed[sce.NeuroD1_CellBender_QCed$scDblFinder.class != "doublet"] sce.NeuroD1_CellBender_QCDbled ``` ``` ## class: SingleCellExperiment ## dim: 37916 1654 ## metadata(3): Samples scDblFinder.stats scDblFinder.threshold ## assays(2): counts logcounts ## rownames(37916): Gm26206 Xkr4 ... MT-TrnT MT-TrnP ## rowData names(4): ID Symbol Type scDblFinder.selected ## colnames(1654): TAGCACAAGACCAACG-1 TGTGCGGGTATGAAGT-1 ... ## GCACGGTCAGGAGACT-1 AAAGAACCATCCTCAC-1 ## colData names(21): Sample Barcode ... scDblFinder.mostLikelyOrigin ## scDblFinder.originAmbiguous ## reducedDimNames(3): PCA TSNE UMAP ## mainExpName: NULL ## altExpNames(0): ``` --- ## Merging 2 Datasets Now we have processed our single dataset we can integrate with our second dataset. I have already processed the Ngn3 dataset in the same way. ``` r plotUMAP(sce.Ngn3_CellBender_QCDbled, colour_by = "All_Cell_Types") ``` <!-- --> --- ## Merging 2 Datasets To merge the two datasets we first need to get objects in sync. Here we make sure the two datasets have the same genes within them ``` r uni <- intersect(rownames(sce.NeuroD1_CellBender_QCDbled), rownames(sce.Ngn3_CellBender_QCDbled)) sce.NeuroD1 <- sce.NeuroD1_CellBender_QCDbled[uni, ] sce.Ngn3 <- sce.Ngn3_CellBender_QCDbled[uni, ] ``` --- ## Merging 2 Datasets We also tidy up the mcols as well. ``` r mcols(sce.NeuroD1) <- mcols(sce.NeuroD1)[, -4] mcols(sce.Ngn3) <- mcols(sce.Ngn3)[, -4] ``` --- ## Merging 2 Datasets We can combine the two datasets with **cbind** ``` r sce.all <- cbind(sce.NeuroD1, sce.Ngn3) ``` --- ## Merging 2 Datasets We need to also gather a common set of variable genes. ``` r dec.NeuroD1 <- dec.NeuroD1_CellBender_QCed[uni, ] dec.Ngn3 <- dec.Ngn3_CellBender_QCed[uni, ] combined.dec <- combineVar(dec.NeuroD1, dec.Ngn3) chosen.hvgs <- combined.dec$bio > 0 ``` --- ## Running PCA and TSNE ``` r library(scater) set.seed(10101010) sce.all <- runPCA(sce.all, subset_row = chosen.hvgs) sce.all <- runTSNE(sce.all, dimred = "PCA") ``` --- ## Review TSNE ``` r plotTSNE(sce.all, colour_by = "Sample") ``` <!-- --> --- ## Review TSNE ``` r plotTSNE(sce.all, colour_by = "Sample") + facet_wrap(~colour_by) ``` <!-- --> --- ## Review TSNE ``` r plotTSNE(sce.all, colour_by = "All_Cell_Types", shape_by = "Sample") + facet_wrap(~shape_by) ``` <!-- --> --- ## Review TSNE ``` r plotTSNE(sce.all, colour_by = "Sample", shape_by = "All_Cell_Types") + facet_wrap(~shape_by) ``` <!-- --> --- ## MNN batch correction. So far we have applied no batch correction to our data. Here we will apply the fastMNN batch correction from the **batchelor** package ``` r library(batchelor) set.seed(1000101001) sce.all.mnn <- fastMNN(sce.all, batch = sce.all$Sample, d = 50, k = 20, subset.row = chosen.hvgs) ``` --- ## Add Celltype annotation again. ``` r eec_paper_meta <- read.delim("https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/GSE224223_EEC_metadata.csv", sep = ";") nrd1_cells <- as.data.frame(eec_paper_meta) nrd1_cells$X <- gsub("_2$|_1$", "", nrd1_cells$X) sce.all.mnn$All_Cell_Types <- nrd1_cells$All_Cell_Types[match(colnames(sce.all.mnn), nrd1_cells$X, nomatch = NA)] ``` --- ##Clustering ``` r library(bluster) clust.louvain <- clusterCells(sce.all.mnn, use.dimred = "corrected", BLUSPARAM = NNGraphParam(cluster.fun = "louvain", cluster.args = list(resolution = 0.5))) colData(sce.all.mnn)$MNNLabel <- clust.louvain ``` --- ## TSNE and UMAP. We can then use the corrected PCA from fastMNN to make our corrected TSNE and UMAPs ``` r sce.all.mnn <- runTSNE(sce.all.mnn, dimred = "corrected", name = "MNN.TSNE") sce.all.mnn <- runUMAP(sce.all.mnn, dimred = "corrected", name = "MNN.UMAP") ``` --- ## Review TSNE and UMAP ``` r plotUMAP(sce.all.mnn, colour_by = "All_Cell_Types", shape_by = "batch", dimred = "MNN.UMAP") + facet_wrap(~shape_by) ``` <!-- --> --- ## Review TSNE and UMAP ``` r plotUMAP(sce.all.mnn, colour_by = "MNNLabel", shape_by = "batch", dimred = "MNN.UMAP") + facet_wrap(~shape_by) ``` <!-- --> --- ## Review TSNE and UMAP ``` r plotUMAP(sce.all.mnn, colour_by = "batch", shape_by = "All_Cell_Types", dimred = "MNN.UMAP") + facet_wrap(~shape_by) ``` <!-- --> --- ## Harmony correction Another apporach to batch correction is implemented in harmony. ``` r library(harmony) sce.all.harmony <- RunHarmony(sce.all, group.by.vars = "Sample") ``` ``` ## Transposing data matrix ``` ``` ## Initializing state using k-means centroids initialization ``` ``` ## Harmony 1/10 ``` ``` ## Harmony 2/10 ``` ``` ## Harmony converged after 2 iterations ``` --- ## Add cell annotation. ``` r sce.all.harmony$All_Cell_Types <- nrd1_cells$All_Cell_Types[match(colnames(sce.all.harmony), nrd1_cells$X, nomatch = NA)] ``` --- ##Clustering ``` r library(bluster) clust.louvain <- clusterCells(sce.all.harmony, use.dimred = "HARMONY", BLUSPARAM = NNGraphParam(cluster.fun = "louvain", cluster.args = list(resolution = 0.5))) colData(sce.all.harmony)$HARMONYLabel <- clust.louvain ``` --- ## TSNE and UMAP. We can then use the corrected PCA from Harmony to make our corrected TSNE and UMAPs. ``` r plotReducedDim(sce.all.harmony, dimred = "HARMONY", colour_by = "subsets_Mito_percent") ``` <!-- --> --- ## TSNE and UMAP. We can then use the corrected PCA from Harmony to make our corrected TSNE and UMAPs. ``` r sce.all.harmony <- runUMAP(sce.all.harmony, dimred = "HARMONY", name = "HARMONY.UMAP") plotUMAP(sce.all.harmony, colour_by = "HARMONYLabel", dimred = "HARMONY.UMAP", shape_by = "Sample") + facet_wrap(~shape_by) ``` <!-- --> --- ## TSNE and UMAP. ``` r plotUMAP(sce.all.harmony, colour_by = "All_Cell_Types", , dimred = "HARMONY.UMAP", shape_by = "Sample") + facet_wrap(~shape_by) ``` <!-- --> --- ## Seurat We can also use our CellBender output in Seurat. At the moment however we have to import using the **scCustomize** package. ``` r library(Seurat) library(scCustomize) h5file <- "https://rubioinformatics.s3.amazonaws.com/scRNA_graduate/SRR_NeuroD1/outs/cellbender_v0.3.2_filtered.h5" local_h5file <- basename(h5file) download.file(h5file, local_h5file) Neurod1.mat <- Read_CellBender_h5_Mat(file_name = local_h5file) Neurod1.obj <- CreateSeuratObject(Neurod1.mat, project = "Nd1") ``` --- ## Seurat We then process as before to get QC and filter. ``` r mito.genes <- grep("^MT", rownames(Neurod1.obj), value = T) percent.mt <- PercentageFeatureSet(Neurod1.obj, features = mito.genes) Neurod1.obj <- AddMetaData(Neurod1.obj, metadata = percent.mt, col.name = "percent.mt") Neurod1.obj.filt <- subset(Neurod1.obj, subset = nCount_RNA < 125000 & percent.mt < 25 & nFeature_RNA > 200) ``` --- ## Seurat And then normalise, run PCA and generate UMAPs. ``` r Neurod1.obj.filt <- NormalizeData(Neurod1.obj.filt) ``` ``` ## Normalizing layer: counts ``` ``` r Neurod1.obj.filt <- FindVariableFeatures(Neurod1.obj.filt, selection.method = "vst", nfeatures = 3000) ``` ``` ## Finding variable features for layer counts ``` ``` r all.genes <- rownames(Neurod1.obj.filt) Neurod1.obj.filt <- ScaleData(Neurod1.obj.filt, features = all.genes) ``` ``` ## Centering and scaling data matrix ``` ``` r Neurod1.obj.filt <- RunPCA(Neurod1.obj.filt, features = VariableFeatures(object = Neurod1.obj)) ``` ``` ## PC_ 1 ## Positive: Bnip5, Trpm2, Apoa4, Apob, Gm53845, Mxd1, Cyp3a11, Klf4, Slc27a4, Col27a1 ## Gm57596, Rasal2, Prss30, Pmp22, Nrp1, Gm50599, Iqsec1, Slc38a11, Ly6m, Apoc3 ## Grik4, Slfn8, Scarb1, Aqp9, Slc34a2, S100a6, Pwwp3b, Serpina1c, Egr1, Stk32a ## Negative: Rps19, Rps5, Rps18, Eef1b2, Rps8, Rps15a, Rpl32, Rpl13, Rpl18a, Rps20 ## Rpl35, Rplp2, Rpl28, Rps23, Rps3, Rps28, Rps7, Rps4x, Rpl23, Rps12 ## Rps9, Rpl3, Rpl35a, Rps29, Rpl14, Rpl30, Pycard, Nme2, Rps21, Cps1 ## PC_ 2 ## Positive: Ptma, Rps3a1, Tff3, Rpl6, Rpl26, Tmem176a, Gm11992, Arx, Krt18, Rpsa ## Rpl13a, Rflna, Parm1, Rpl3, Rps18, Rpl21, Rpl8, Agr3, Rps7, Gm53467 ## Cltrn, Rpl13, Rpl11, Etv1, Rps21, Rpl5, Rpl17, Anp32b, Kcnk16, Rpl32 ## Negative: Spink1, Clec2h, Mep1b, Guca2b, Ces2e, Ace2, Chp2, Slc51a, Lct, Ggt1 ## Clca4b, Treh, Alpi, Apoa4, Ace, Apol10a, Xdh, H2-T26, Cndp2, Btnl1 ## Tm6sf2, Maf, Ifi27l2b, St3gal4, Arg2, H2-T3, Maoa, Tgfbi, Pls1, Dnase1 ## PC_ 3 ## Positive: Arx, Cyp2j6, Gm11992, Rflna, Etv1, Sult1d1, Pzp, Cltrn, Parm1, Kcnk16 ## Ghrl, Crp, Galnt13, S100a11, Gnai1, Ugt2b34, Isl1, Tril, Gm53467, Smarca1 ## Adamts5, Tmem176a, Dner, Tle1, Casr, Slc41a2, Basp1, Ugt8a, Gsto1, Tlr2 ## Negative: Ccnd2, Krt19, Slc38a11, Tpbg, Rasal2, Trpm2, Sorl1, Gm53845, Amigo2, Fmo2 ## Igfbp4, Iqsec1, Serpinf2, Grik4, Aqp9, 9330154J02Rik, Tac1, Nars2, Slc5a9, Tmem238l ## Rab3c, Cartpt, Apoc3, Tnfrsf11b, Slc18a2, Acsl6, Fabp3, Serpini1, S100a6, Acvrl1 ## PC_ 4 ## Positive: Top2a, Cenpf, Pbk, Nusap1, Pclaf, D17H6S56E-5, Tpx2, Cenpe, H1f5, Ckap2l ## Birc5, Kif23, Gvin-ps1, Clca3b, Cdk1, Gip, Sgo1, Hmgb2, Aurkb, Mis18bp1 ## Melk, Cdca2, Ccna2, Cks2, Prc1, Uhrf1, Tk1, Bub1, Cenpw, Ncaph ## Negative: Tff3, Serpinf2, Slc38a11, Ctsl, Apoc3, Krt19, Atp5f1c, Agr3, Upb1, Uqcr10 ## Uqcr11, Tpbg, Ppia, Phgr1, Fmo2, Sorl1, Ttyh1, Ccnd2, Ndufa4, Fabp3 ## Rab3c, Rasal2, Atp5mf, Uqcrh, Pdzk1, Uqcrb, Atp5mj, Akr1c14, Fth1, Clps ## PC_ 5 ## Positive: Gip, Mrln, Scarb1, Sphkap, Rgs4, Pkib, Rbp2, Ass1, Trpc5, Pwwp3b ## Slfn10-ps, Fmo1, Vim, Th, Isl1, Cth, Pls3, Dnah9, Gm32255, Cox7a2l ## Gsto1, Prps1, Bdnf, Gm54263, 1700086L19Rik, Acvr1c, Hpd, Slfn8, Acvrl1, Inhbb ## Negative: Agr2, Agr3, Phgr1, Onecut3, Pzp, Ret, Homer2, Tle1, Casr, Galnt13 ## Crp, Serpina1c, Ppic, Gm12511, Serpina1b, Cit, Klf4, Hspb1, Ckb, Kcnk16 ## S100a10, Gm53467, Slc41a2, Adamts5, Fgl2, Basp1, Glod5, Ugt8a, Serpina1d, S100a6 ``` ``` r Neurod1.obj.filt <- RunUMAP(Neurod1.obj.filt, dims = 1:30) ``` ``` ## 11:24:08 UMAP embedding parameters a = 0.9922 b = 1.112 ``` ``` ## 11:24:08 Read 1653 rows and found 30 numeric columns ``` ``` ## 11:24:08 Using Annoy for neighbor search, n_neighbors = 30 ``` ``` ## 11:24:08 Building Annoy index with metric = cosine, n_trees = 50 ``` ``` ## 0% 10 20 30 40 50 60 70 80 90 100% ``` ``` ## [----|----|----|----|----|----|----|----|----|----| ``` ``` ## **************************************************| ## 11:24:08 Writing NN index file to temp file /var/folders/0_/8lnwn5sn7hn9nr8qv2jg48nw0000gn/T//RtmpUTDURW/file16eb7ccd0749 ## 11:24:08 Searching Annoy index using 1 thread, search_k = 3000 ## 11:24:09 Annoy recall = 100% ## 11:24:10 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30 ## 11:24:12 Initializing from normalized Laplacian + noise (using RSpectra) ## 11:24:12 Commencing optimization for 500 epochs, with 65124 positive edges ## 11:24:15 Optimization finished ``` --- ## Seurat And finally define our clusters ``` r Neurod1.obj.filt <- FindNeighbors(Neurod1.obj.filt, dims = 1:30) ``` ``` ## Computing nearest neighbor graph ``` ``` ## Computing SNN ``` ``` r Neurod1.obj.filt <- FindClusters(Neurod1.obj.filt, resolution = 0.7) ``` ``` ## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck ## ## Number of nodes: 1653 ## Number of edges: 53942 ## ## Running Louvain algorithm... ## Maximum modularity in 10 random starts: 0.8606 ## Number of communities: 11 ## Elapsed time: 0 seconds ``` --- ## Seurat Now we can review our UMAP ``` r DimPlot(Neurod1.obj.filt, reduction = "umap") ``` <!-- --> --- ## Ngn3 Seurat ``` r DimPlot(Ngn3.obj.filt, reduction = "umap") ``` <!-- --> --- ## Seurat- 2 datasets We can combine two datasets using the merge function in Seurat. ``` r All.obj <- merge(Neurod1.obj.filt, y = Ngn3.obj.filt, add.cell.ids = c("Nd1", "Ngn3"), project = "All", merge.data = TRUE) All.obj ``` ``` ## An object of class Seurat ## 39905 features across 8832 samples within 1 assay ## Active assay: RNA (39905 features, 3000 variable features) ## 6 layers present: counts.Nd1, counts.Ngn3, data.Nd1, scale.data.Nd1, data.Ngn3, scale.data.Ngn3 ``` --- ## Seurat- 2 datasets Once merged we can run a standard processing. ``` r All.obj <- NormalizeData(All.obj) ``` ``` ## Normalizing layer: counts.Nd1 ``` ``` ## Normalizing layer: counts.Ngn3 ``` ``` r All.obj <- FindVariableFeatures(All.obj) ``` ``` ## Finding variable features for layer counts.Nd1 ``` ``` ## Finding variable features for layer counts.Ngn3 ``` ``` r All.obj <- ScaleData(All.obj) ``` ``` ## Centering and scaling data matrix ``` ``` r All.obj <- RunPCA(All.obj) ``` ``` ## PC_ 1 ## Positive: Tm4sf4, Selenop, Sct, Krt20, Syt7, Neurod1, Cldn4, Adgrg4, Gpx3, Cdkn1a ## Peg3, Rgs2, Prnp, Pcsk1, Resp18, Cacna2d1, Scg5, Rundc3a, Ambp, Mxd1 ## Cpe, Stxbp5l, Kcnb2, Pcsk1n, Aplp1, Celf3, Chgb, Scg3, Tph1, Ttyh1 ## Negative: Gvin-ps1, Hspe1, Rps2, C1qbp, Hspd1, Pycard, Aldh1b1, Rps11, Rps19, Rps4x ## Cps1, Rps5, Nme2, Eef1b2, Rpl12, Rps8, Rpl23a, Csrp2, Hmgb2, Rplp0 ## Rps18, Rpl7, Snrpg, Ran, Rps16, Rpl22, Oat, Rps12, Rpl27a, Rps25 ## PC_ 2 ## Positive: Mep1b, Mttp, Ces2e, Slc26a6, Sult1b1, Spink1, Mogat2, Cyp4f14, Slc51a, Chp2 ## 2200002D01Rik, Ace2, Enpep, Ckmt1, Tm6sf2, Anpep, Dgat1, St3gal4, Arg2, Khk ## Rbp2, Adh6a, Fabp2, Il18, Gstm3, Gda, Fabp1, Acsl5, Cndp2, Mpp1 ## Negative: Olfm4, Mmp7, Defa42, Lyz1, Defa39, Defa26, Itln1, Defa17, Defa38, Defa29 ## Ifitm3, Defa22, Mptx2, Defa21, Defa5, Defa40, Slc12a2, Ang4, Cd24a, Clps ## Pnliprp2, Defa3, Tubb5, Hmgn1, Defa37, Rnase1, Defa28, Defa24, Stmn1, Defa34 ## PC_ 3 ## Positive: Chgb, Neurod1, Pcsk1, Prnp, Cpe, Ddc, Scg5, Bex3, Aplp1, Resp18 ## Chga, Pam, Gpx3, Cacna2d1, Cfap144, Adgrg4, Bex2, Selenop, Scg3, Ambp ## Tph1, Peg3, Sct, Celf3, Slc18a1, Cacna1a, Pcsk1n, Insm1, Scn3a, Kcnb2 ## Negative: Spink4, Muc2, Ccl6, Zg16, Clca1, Itln1, Defa24, Nupr1, Ang4, Clps ## Tspan8, Lyz1, Lrrc26, Fcgbp, Guca2a, Pglyrp1, Defa17, Mmp7, Ano7, Defa39 ## Fer1l6, Defa42, Sytl2, Defa26, Defa29, Defa38, Klk1, Defa5, Defa22, Defa21 ## PC_ 4 ## Positive: Hck, Sh2d6, Alox5ap, Rgs13, Irag2, Dclk1, Avil, Nrgn, Ptgs1, Trpm5 ## Siglecf, Sh2d7, Ltc4s, Spib, Matk, Fyb1, Bmx, Hmx2, Vav1, Alox5 ## Ly6g6d, Smpx, Nebl, Tspan6, Pik3r5, Cd300lf, Strip2, Hebp1, Ccnj, Rac2 ## Negative: Spink4, Clps, Itln1, Guca2b, Lypd8l, Lyz1, Reg4, Ccl6, Ang4, Defa39 ## Fabp2, Defa17, Mmp7, Defa42, Fos, Defa24, Pglyrp1, Fcgbp, Agr2, Nupr1 ## Defa21, Defa22, Prap1, Defa38, Muc2, Defa26, Gpx2, Aldob, Ccnd2, Reg3g ## PC_ 5 ## Positive: Ccnd2, Fcgbp, S100a6, Clca1, AW112010, Fer1l6, Krt18, Slc38a11, Ano7, Tpbg ## Afp, Gsn, Dlg2, 2810025M15Rik, Rasal2, Lmx1a, Bcas1, Tpsg1, Sytl2, Klk1 ## Muc2, Kpna2, Lypd8, Trpm2, Capn9, Zg16, Trpa1, Ifi27l2b, Ido1, Atoh1 ## Negative: Rbp4, Scg2, Cck, Scgn, Bambi, Isl1, Arx, Gm11992, Fabp5, Etv1 ## Rflna, Defa26, Defa29, Defa42, Mptx2, Abcc8, Defa38, Cltrn, Defa40, Defa5 ## Defa39, Ghrl, Habp2, Defa21, Defa22, Gip, Defa17, Sphkap, Gnai1, Kcnk16 ``` ``` r All.obj <- FindNeighbors(All.obj, dims = 1:30, reduction = "pca") ``` ``` ## Computing nearest neighbor graph ``` ``` ## Computing SNN ``` ``` r All.obj <- FindClusters(All.obj, resolution = 2, cluster.name = "unintegrated_clusters") ``` ``` ## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck ## ## Number of nodes: 8832 ## Number of edges: 282167 ## ## Running Louvain algorithm... ## Maximum modularity in 10 random starts: 0.8450 ## Number of communities: 37 ## Elapsed time: 0 seconds ``` ``` r All.obj <- RunUMAP(All.obj, dims = 1:30, reduction = "pca", reduction.name = "umap.unintegrated") ``` ``` ## 11:26:12 UMAP embedding parameters a = 0.9922 b = 1.112 ``` ``` ## 11:26:12 Read 8832 rows and found 30 numeric columns ``` ``` ## 11:26:12 Using Annoy for neighbor search, n_neighbors = 30 ``` ``` ## 11:26:12 Building Annoy index with metric = cosine, n_trees = 50 ``` ``` ## 0% 10 20 30 40 50 60 70 80 90 100% ``` ``` ## [----|----|----|----|----|----|----|----|----|----| ``` ``` ## **************************************************| ## 11:26:13 Writing NN index file to temp file /var/folders/0_/8lnwn5sn7hn9nr8qv2jg48nw0000gn/T//RtmpUTDURW/file16eb6754ab37 ## 11:26:13 Searching Annoy index using 1 thread, search_k = 3000 ## 11:26:16 Annoy recall = 100% ## 11:26:17 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30 ## 11:26:19 Initializing from normalized Laplacian + noise (using RSpectra) ## 11:26:19 Commencing optimization for 500 epochs, with 363508 positive edges ## 11:26:31 Optimization finished ``` --- ## Seurat- Review merging We can review any batch effects using our umap. ``` r DimPlot(All.obj, reduction = "umap.unintegrated", group.by = "orig.ident") ``` <!-- --> --- ## Seurat- Integration We can integrate and batch correct our datasets using the **IntegrateLayers** layers function. This allows for many different integration methods. Here we will use Harmony again. ``` r All.obj <- IntegrateLayers(object = All.obj, method = HarmonyIntegration, orig.reduction = "pca", new.reduction = "integrated.harmony", verbose = TRUE) ``` ``` ## Transposing data matrix ``` ``` ## Using automatic lambda estimation ``` ``` ## Initializing state using k-means centroids initialization ``` ``` ## Harmony 1/10 ``` ``` ## Harmony 2/10 ``` ``` ## Harmony 3/10 ``` ``` ## Harmony converged after 3 iterations ``` --- ## Seurat- Downstream of integration Now we remake our clusters using the harmony integrated data. ``` r All.obj[["RNA"]] <- JoinLayers(All.obj[["RNA"]]) All.obj <- FindNeighbors(All.obj, reduction = "integrated.harmony", dims = 1:30) ``` ``` ## Computing nearest neighbor graph ``` ``` ## Computing SNN ``` ``` r All.obj <- FindClusters(All.obj, resolution = 0.5, cluster.name = "harmony_clusters") ``` ``` ## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck ## ## Number of nodes: 8832 ## Number of edges: 284961 ## ## Running Louvain algorithm... ## Maximum modularity in 10 random starts: 0.9246 ## Number of communities: 17 ## Elapsed time: 0 seconds ``` --- ## Seurat- Downstream of integration Lastly we can create our UMAP from harmony integrated data. ``` r All.obj <- RunUMAP(All.obj, reduction = "integrated.harmony", dims = 1:30, reduction.name = "umap.harmony") ``` ``` ## 11:26:56 UMAP embedding parameters a = 0.9922 b = 1.112 ``` ``` ## 11:26:56 Read 8832 rows and found 30 numeric columns ``` ``` ## 11:26:56 Using Annoy for neighbor search, n_neighbors = 30 ``` ``` ## 11:26:56 Building Annoy index with metric = cosine, n_trees = 50 ``` ``` ## 0% 10 20 30 40 50 60 70 80 90 100% ``` ``` ## [----|----|----|----|----|----|----|----|----|----| ``` ``` ## **************************************************| ## 11:26:57 Writing NN index file to temp file /var/folders/0_/8lnwn5sn7hn9nr8qv2jg48nw0000gn/T//RtmpUTDURW/file16eb17f76071 ## 11:26:57 Searching Annoy index using 1 thread, search_k = 3000 ## 11:27:00 Annoy recall = 100% ## 11:27:01 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30 ## 11:27:03 Initializing from normalized Laplacian + noise (using RSpectra) ## 11:27:03 Commencing optimization for 500 epochs, with 364320 positive edges ## 11:27:15 Optimization finished ``` --- ## Seurat- Review integration ``` r DimPlot(All.obj, reduction = "umap.harmony", group.by = "harmony_clusters", combine = FALSE, label.size = 2) ``` ``` ## [[1]] ``` <!-- --> --- ## Seurat- Review integration ``` r DimPlot(All.obj, reduction = "umap.harmony", group.by = "orig.ident", combine = FALSE, label.size = 2) ``` ``` ## [[1]] ``` <!-- --> --- ## Markers within/between batches Batch correction is often performed on the PCs (MNN and Harmony) and so is used for UMAP and clustering. ``` r plotUMAP(sce.all.mnn, colour_by = "MNNLabel", dimred = "MNN.UMAP") ``` <!-- --> --- ## Markers within/between batches MNN correction does return a batch corrected matrix (**reconstructed** assay) but this is not recommended for differential expression or marker gene detection. Instead it is recommended we use the log normalised data and account for batches as part of our marker gene detection. First then we add MNN clusters detected back to our uncorrected SingleCellExperiment object. ``` r sce.all$MNNLabel <- sce.all.mnn$MNNLabel sce.all$batch <- sce.all.mnn$batch ``` --- ## Markers within/between batches We now run the findMarkers function adding an additional parameter of **block** to identify markers of MNN identified clusters while accounting for batch/sample differences. ``` r m.out <- findMarkers(sce.all, sce.all$MNNLabel, block = sce.all$batch, direction = "up", lfc = 1) ``` --- ## Markers within/between batches We can then review these as before ``` r m.out[["1"]][, c("summary.logFC", "Top", "p.value", "FDR")] ``` ``` ## DataFrame with 33650 rows and 4 columns ## summary.logFC Top p.value FDR ## <numeric> <integer> <numeric> <numeric> ## Hspe1 4.67793 1 0.00000e+00 0.00000e+00 ## Tm4sf20 3.19782 1 0.00000e+00 0.00000e+00 ## Aldh1b1 3.81728 1 0.00000e+00 0.00000e+00 ## Reg3b 3.47170 1 1.36616e-92 6.28022e-91 ## Rps17 5.00060 1 0.00000e+00 0.00000e+00 ## ... ... ... ... ... ## Gm21748 -0.00100595 33626 1 1 ## LOC100861691 0.00000000 33627 1 1 ## MT-TrnL1 0.00378810 33633 1 1 ## MT-TrnK -0.00684009 33643 1 1 ## MT-TrnH -0.00677348 33646 1 1 ``` --- ## Markers within/between batches And visualise these markers in clusters while splitting by batch/sample. ``` r plotExpression(sce.all, features = c("Chgb", "Reg4"), x = "MNNLabel", colour_by = "batch") + facet_wrap(Feature ~ colour_by) ``` <!-- --> --- ## Markers within/between batches We can also visualise them using the MNN **reconstructed** matrix. ``` r plotExpression(sce.all.mnn, features = c("Chgb", "Reg4"), x = "MNNLabel", exprs_values = "reconstructed", colour_by = "batch") + facet_wrap(Feature ~ colour_by) ``` <!-- --> --- ## Markers within/between batches Finding markers within a cluster across samples/condition while accounting for batch is not possible given that batch is the same as sample/condition. We can however combine cluster/sample labels and make a comparison across conditions. ``` r sce.all$Condition_Cluster <- paste(sce.all$Sample, sce.all$MNNLabel, sep = "_") Nd1_vs_Ngn3_Cluster9 <- scoreMarkers(sce.all, sce.all$Condition_Cluster, pairings = c("Nd1_9", "Ngn3_9")) ``` --- ## Markers within/between batches Reviewing this we see that we are now simply capturing difference between samples. ``` r Nd1_vs_Ngn3_Cluster9_Res <- Nd1_vs_Ngn3_Cluster9$Nd1_9 Nd1_vs_Ngn3_Cluster9_Res <- Nd1_vs_Ngn3_Cluster9_Res[order(Nd1_vs_Ngn3_Cluster9_Res$mean.AUC, decreasing = TRUE), ] plotExpression(sce.all, features = c("Xist"), x = "MNNLabel", colour_by = "batch") + facet_wrap(Feature ~ colour_by) ``` <!-- --> --- ## Seurat Seurat also offers a mechanism to block with in batches for identifying cluster markers while accounting for samples. ``` r Idents(All.obj) <- "harmony_clusters" harmony.markers <- FindConservedMarkers(All.obj, ident.1 = 1, grouping.var = "orig.ident", verbose = FALSE) VlnPlot(All.obj, features = "Slc38a11", split.by = "orig.ident") ``` <!-- --> --- ## Seurat However we have the same issue for when comparing across conditions. ``` r All.obj[["Condition_Cluster"]] <- paste(All.obj$orig.ident, All.obj$harmony_clusters, sep = "_") Idents(All.obj) <- "Condition_Cluster" res <- FindMarkers(All.obj, ident.1 = "Nd1_1", ident.2 = "Ngn3_1", verbose = FALSE) ``` --- ## scTransform. scTransform is an alternative workflow for normalising and scaling data which was put forward by Seurat authors. It replaces normalised counts with residuals from fitting a model per gene while also allowing us to regress out features from our data. ``` r Neurod1.obj.scTransform <- subset(Neurod1.obj, subset = nCount_RNA < 125000 & percent.mt < 25 & nFeature_RNA > 200) Neurod1.obj.scTransform <- SCTransform(Neurod1.obj.scTransform, vars.to.regress = "percent.mt") ``` ``` ## Running SCTransform on assay: RNA ``` ``` ## Running SCTransform on layer: counts ``` ``` ## vst.flavor='v2' set. Using model with fixed slope and excluding poisson genes. ``` ``` ## Variance stabilizing transformation of count matrix of size 17675 by 1653 ``` ``` ## Model formula is y ~ log_umi ``` ``` ## Get Negative Binomial regression parameters per gene ``` ``` ## Using 2000 genes, 1653 cells ``` ``` ## Found 36 outliers - those will be ignored in fitting/regularization step ``` ``` ## Second step: Get residuals using fitted parameters for 17675 genes ``` ``` ## Computing corrected count matrix for 17675 genes ``` ``` ## Calculating gene attributes ``` ``` ## Wall clock passed: Time difference of 11.93546 secs ``` ``` ## Determine variable features ``` ``` ## Regressing out percent.mt ``` ``` ## Centering data matrix ``` ``` ## Set default assay to SCT ``` --- ## scTransform. An additional SCT slot is created containing these residuals for further analysis. ``` r Neurod1.obj.scTransform ``` ``` ## An object of class Seurat ## 57580 features across 1653 samples within 2 assays ## Active assay: SCT (17675 features, 3000 variable features) ## 3 layers present: counts, data, scale.data ## 1 other assay present: RNA ``` --- ## scTransform. Following this transformation we can procede as normal with analysis. ``` r Neurod1.obj.scTransform <- RunPCA(Neurod1.obj.scTransform) ``` ``` ## PC_ 1 ## Positive: Chga, Reg4, Tph1, Slc38a11, Afp, Ccnd2, Krt19, Dlg2, Tpbg, Krt7 ## Trpm2, Ambp, Rasal2, Gpx3, Itprid2, 2810025M15Rik, Apoc3, Cartpt, Chgb, Trpa1 ## Slc18a1, Stxbp5l, Lmx1a, Serpina1c, Lypd8l, Pla2g7, Rgs2, Shisa9, Apoa1, Cwh43 ## Negative: Cck, Scg2, Rbp4, Ghrl, Fabp5, Rps24, Rps4x, Rps18, Rpl32, Rps2 ## Rps19, Cps1, Rps20, Rpl12, Rpl13, Rpl23, Pycard, Rplp0, Ifitm2, Gpx2 ## Rps15a, Rpl27a, Aldh1b1, Rps16, Tspan8, Isl1, Gip, Rpl39, Rps8, Cd74 ## PC_ 2 ## Positive: Scg2, Cck, Rbp4, Sct, Ghrl, Isl1, Fabp5, Nts, Gip, Myl7 ## Scgn, Etv1, Cpe, Bambi, Rgs4, Rflna, Abcc8, Pzp, Agr2, Bace2 ## Sphkap, Cnr1, Slc8a1, Upp1, Phlda1, Arx, Cyp2j6, Gm11992, Cltrn, Onecut3 ## Negative: Krt19, Reg4, Tmsb4x, Chga, Gvin-ps1, Ccnd2, Rps19, Gpx2, Pycard, Aldh1b1 ## Rps12, Rps11, Rplp1, Hspe1, Rps8, Rplp0, Rps2, Tspan8, Mgst1, Cps1 ## Mt1, Rpl13, Oat, Ppp1r1b, Rpl28, Afp, Rps5, Clca3b, Rpl35, Dmbt1 ## PC_ 3 ## Positive: Spink1, Apoa1, Clec2h, Apoa4, 2200002D01Rik, Fabp2, Gda, Guca2b, Mttp, St3gal4 ## Prap1, Lgals3, Ace2, Ggt1, Aldob, Dgat1, Clca4b, Ifi27l2b, Mep1b, Ces2e ## Alpi, Xdh, Slc5a1, Fabp1, Sult1b1, Rbp2, 2010106E10Rik, Chp2, Slc26a6, Anpep ## Negative: Clca3b, Slc12a2, Ncl, Olfm4, Npm1, Rpsa, Rpl12, Stmn1, Gas5, Xist ## Rplp1, Rps18, Ifitm2, Gvin-ps1, Ifitm3, Rps12, Rpl27a, Tpbg, Rpl13, Rpl32 ## D17H6S56E-5, Chga, Ptma, Rps20, Scn3a, Rpl35, Impdh2, Hmgb2, Rps24, Rpl3 ## PC_ 4 ## Positive: Agr2, Agr3, Pzp, Tff3, Onecut3, Cck, Habp2, Lgals2, Mgll, Sct ## Ghrl, Cyp2j5, Krt20, Serpina1e, Cyp2j6, Gsdma, Slc39a8, Pgc, Ms4a8a, Scg2 ## Basp1, Tle1, Adamts5, Crp, Etv1, Fcgbp, Rgs2, Serpina1b, Kcnk16, Tph1 ## Negative: Gip, Rgs4, Fabp5, Phlda1, Cd44, Cnr1, Fxyd6, Rbp2, Isl1, Sphkap ## Pkib, Rfx6, Itga4, Scarb1, Gatm, Car8, Slfn10-ps, 2010204K13Rik, Pwwp3b, Entpd5 ## Th, Mrln, Abcc8, Fam167a, Vim, Prxl2a, Entpd3, Plcxd3, Meg3, Amigo2 ## PC_ 5 ## Positive: Tff3, Afp, Pzp, Trpa1, Cck, Gstt1, Amigo2, Akr1c14, Gstk1, Slc5a9 ## 2810025M15Rik, Ucn3, Habp2, Scg2, Ptgr1, Cyp2j5, Rgs2, Pla2g7, Tm4sf5, Tpbg ## Hip1, Rasal2, Pigr, Fabp1, Slc13a1, Pgc, Slc18a2, Fabp3, Hibadh, Cyp2j6 ## Negative: Cd24a, Serpina1c, Serpina1e, Syt7, Hspb1, S100a10, Gm32585, S100a6, Trpm2, Krt7 ## Cartpt, Fos, Col27a1, Wif1, Tgfb1, Tmsb4x, Pcsk1n, Serpina1a, Rgs4, Gip ## Jun, Pyy, Prss30, Galr1, Mgll, Espn, Slc15a1, Egr1, Klf4, Cldn4 ``` ``` r Neurod1.obj.scTransform <- RunUMAP(Neurod1.obj.scTransform, dims = 1:30) ``` ``` ## 11:30:38 UMAP embedding parameters a = 0.9922 b = 1.112 ``` ``` ## 11:30:38 Read 1653 rows and found 30 numeric columns ``` ``` ## 11:30:38 Using Annoy for neighbor search, n_neighbors = 30 ``` ``` ## 11:30:38 Building Annoy index with metric = cosine, n_trees = 50 ``` ``` ## 0% 10 20 30 40 50 60 70 80 90 100% ``` ``` ## [----|----|----|----|----|----|----|----|----|----| ``` ``` ## **************************************************| ## 11:30:38 Writing NN index file to temp file /var/folders/0_/8lnwn5sn7hn9nr8qv2jg48nw0000gn/T//RtmpUTDURW/file16eb2582ef5c ## 11:30:38 Searching Annoy index using 1 thread, search_k = 3000 ## 11:30:38 Annoy recall = 100% ## 11:30:39 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30 ## 11:30:42 Initializing from normalized Laplacian + noise (using RSpectra) ## 11:30:42 Commencing optimization for 500 epochs, with 64510 positive edges ## 11:30:45 Optimization finished ``` ``` r Neurod1.obj.scTransform <- FindNeighbors(Neurod1.obj.scTransform, dims = 1:30) ``` ``` ## Computing nearest neighbor graph ## Computing SNN ``` ``` r Neurod1.obj.scTransform <- FindClusters(Neurod1.obj.scTransform, resolution = 0.7) ``` ``` ## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck ## ## Number of nodes: 1653 ## Number of edges: 50991 ## ## Running Louvain algorithm... ## Maximum modularity in 10 random starts: 0.8589 ## Number of communities: 12 ## Elapsed time: 0 seconds ``` --- ## scTransform. ``` ## An object of class Seurat ## 59126 features across 7179 samples within 2 assays ## Active assay: SCT (19221 features, 3000 variable features) ## 3 layers present: counts, data, scale.data ## 1 other assay present: RNA ## 2 dimensional reductions calculated: pca, umap ``` We can also use scTransform with Batch correction. First we must prepare and merge our data. ``` r All.obj.sct <- merge(Neurod1.obj.scTransform, y = Ngn3.obj.scTransform, add.cell.ids = c("Nd1", "Ngn3"), project = "All", merge.data = TRUE) All.obj.sct <- SCTransform(All.obj.sct, vars.to.regress = "percent.mt") ``` ``` ## Running SCTransform on assay: RNA ``` ``` ## Running SCTransform on layer: counts.Nd1 ``` ``` ## vst.flavor='v2' set. Using model with fixed slope and excluding poisson genes. ``` ``` ## Variance stabilizing transformation of count matrix of size 17675 by 1653 ``` ``` ## Model formula is y ~ log_umi ``` ``` ## Get Negative Binomial regression parameters per gene ``` ``` ## Using 2000 genes, 1653 cells ``` ``` ## Found 36 outliers - those will be ignored in fitting/regularization step ``` ``` ## Second step: Get residuals using fitted parameters for 17675 genes ``` ``` ## Computing corrected count matrix for 17675 genes ``` ``` ## Calculating gene attributes ``` ``` ## Wall clock passed: Time difference of 13.19651 secs ``` ``` ## Determine variable features ``` ``` ## Regressing out percent.mt ``` ``` ## Centering data matrix ``` ``` ## Running SCTransform on layer: counts.Ngn3 ``` ``` ## vst.flavor='v2' set. Using model with fixed slope and excluding poisson genes. ``` ``` ## Variance stabilizing transformation of count matrix of size 19221 by 7179 ``` ``` ## Model formula is y ~ log_umi ``` ``` ## Get Negative Binomial regression parameters per gene ``` ``` ## Using 2000 genes, 5000 cells ``` ``` ## Found 126 outliers - those will be ignored in fitting/regularization step ``` ``` ## Second step: Get residuals using fitted parameters for 19221 genes ``` ``` ## Computing corrected count matrix for 19221 genes ``` ``` ## Calculating gene attributes ``` ``` ## Wall clock passed: Time difference of 42.66531 secs ``` ``` ## Determine variable features ``` ``` ## Regressing out percent.mt ``` ``` ## Centering data matrix ``` ``` ## Getting residuals for block 1(of 2) for Ngn3 dataset ``` ``` ## Getting residuals for block 2(of 2) for Ngn3 dataset ``` ``` ## Regressing out percent.mt ``` ``` ## Centering data matrix ``` ``` ## Finished calculating residuals for Ngn3 ``` ``` ## Regressing out percent.mt ``` ``` ## Centering data matrix ``` ``` ## Regressing out percent.mt ``` ``` ## Centering data matrix ``` ``` ## Set default assay to SCT ``` --- ## scTransform. Following this we an integrate our data using the SCT normalised values. We simply specify "SCT" to the normalisation.method parameter. ``` r All.obj.sct <- RunPCA(All.obj.sct) ``` ``` ## PC_ 1 ## Positive: Apoa1, Rbp2, Fabp1, Spink1, Sis, Apoc3, Selenop, Clec2h, Apoa4, Gsta1 ## Mep1b, Krt20, Gda, Fabp2, Adh6a, St3gal4, Anpep, Tph1, Reg3b, Chga ## Ace2, Sct, Mttp, Chgb, Ggt1, Papss2, Cyp4f14, Ces2e, Apob, Gpx3 ## Negative: Defa24, Lyz1, Itln1, Olfm4, Ang4, Defa17, Mmp7, Clps, Pnliprp2, Ifitm3 ## Rnase1, Stmn1, Clca3b, Hmgb2, Gvin-ps1, Slc12a2, H2ac23, Top2a, Ube2c, Defa37 ## Tubb5, Npm1, Pclaf, Cps1, Tuba1b, D17H6S56E-5, Rps19, Mki67, Rps2, Rpsa ## PC_ 2 ## Positive: Chgb, Chga, Tph1, Sct, Neurod1, Gpx3, Reg4, Ambp, Cpe, Selenop ## Afp, Pcsk1, Myl7, Rgs2, Peg3, Resp18, Pcsk1n, Slc38a11, Krt7, Slc18a1 ## Aplp1, Prnp, Scn3a, Scg5, Syt7, Tm4sf4, Tac1, Stxbp5l, Adgrg4, 2810025M15Rik ## Negative: Reg3b, St3gal4, Spink1, Clec2h, Reg3g, Gsta1, Fabp2, Mep1b, Arg2, Ces2e ## Mttp, Reg1, Anpep, Zg16, Rbp2, Fabp1, Adh6a, Gda, Gstm3, Ace2 ## Cyp4f14, Sult1b1, Il18, Guca2b, Slc5a1, Defa24, Slc51a, Mogat2, 2200002D01Rik, Ggt1 ## PC_ 3 ## Positive: Zg16, Tff3, Fcgbp, Clca1, Muc2, AW112010, Agr2, Spink4, Klk1, Ccl6 ## Fer1l6, Gsta4, Guca2a, Ido1, Tpsg1, Ifi27l2b, Mxd1, Sytl2, Ano7, Lrrc26 ## Klf4, Capn9, Gcnt3, S100a6, Bcas1, Gsn, Krt20, Neat1, Pla2g10, Tm4sf4 ## Negative: Fabp1, Chgb, Olfm4, Rbp2, Reg3b, Adh6a, Reg1, Sis, Chga, Prap1 ## Arg2, Ces2e, Dmbt1, Mttp, Aldob, Apoa1, Anpep, Gstm3, Ccl25, Mep1b ## Apoc3, Spink1, Ckb, Sult1b1, Maoa, Reg4, Khk, Ifitm3, Slc5a1, Mogat2 ## PC_ 4 ## Positive: Defa24, Lyz1, Itln1, Ang4, Defa17, Clps, Pnliprp2, Mmp7, Defa37, Rnase1 ## Reg4, Guca2b, Apoa1, Nupr1, Defa41, Apoa4, Apoc3, Tmed6, Ggt1, Gda ## Sis, Selenop, Habp2, Cyp3a11, Apob, Ace2, Sct, Rbp2, Slc5a1, Bambi ## Negative: Dmbt1, Hmgb2, Clca1, Gvin-ps1, Ube2c, Fcgbp, H2ac23, Zg16, Top2a, Pycard ## Tff3, Agr2, Birc5, Stmn1, Pclaf, Mki67, Muc2, Tuba1b, Cps1, Rps2 ## Cenpf, Hspd1, Cdca3, C1qbp, Tubb5, Tmsb4x, Hspe1, Aldh1b1, Clca3b, D17H6S56E-5 ## PC_ 5 ## Positive: Cck, Rbp4, Scg2, Ghrl, Kctd12, Sh2d6, Dclk1, Fyb1, Hck, Irag2 ## Avil, Tm4sf4, Alox5ap, Trpm5, Ltc4s, Tmem176a, Tuba1a, Snrnp25, Cd24a, Matk ## Rac2, Ptpn18, Nrgn, Ptpn6, Basp1, Espn, Aldh2, Tmem176b, Fabp5, Tspan6 ## Negative: Reg4, Chga, Afp, Tph1, Ccnd2, Trpa1, Slc38a11, Fcgbp, 2810025M15Rik, Tpbg ## Tac1, Krt19, Apoc3, Rasal2, Cartpt, Dlg2, Zg16, Krt7, Amigo2, Clps ## Apoa1, Clca1, Lypd8l, Spink4, Gstt1, Tff3, Muc2, Slc18a1, Pla2g7, Ambp ``` ``` r All.obj.sct <- RunUMAP(All.obj.sct, dims = 1:30) ``` ``` ## 11:35:04 UMAP embedding parameters a = 0.9922 b = 1.112 ``` ``` ## 11:35:04 Read 8832 rows and found 30 numeric columns ``` ``` ## 11:35:04 Using Annoy for neighbor search, n_neighbors = 30 ``` ``` ## 11:35:04 Building Annoy index with metric = cosine, n_trees = 50 ``` ``` ## 0% 10 20 30 40 50 60 70 80 90 100% ``` ``` ## [----|----|----|----|----|----|----|----|----|----| ``` ``` ## **************************************************| ## 11:35:05 Writing NN index file to temp file /var/folders/0_/8lnwn5sn7hn9nr8qv2jg48nw0000gn/T//RtmpUTDURW/file16eb6ff3f844 ## 11:35:05 Searching Annoy index using 1 thread, search_k = 3000 ## 11:35:07 Annoy recall = 100% ## 11:35:08 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30 ## 11:35:11 Initializing from normalized Laplacian + noise (using RSpectra) ## 11:35:11 Commencing optimization for 500 epochs, with 358442 positive edges ## 11:35:22 Optimization finished ``` ``` r All.obj.sct <- IntegrateLayers(object = All.obj.sct, method = HarmonyIntegration, normalization.method = "SCT", verbose = F) ``` --- ## scTransform. Once transformed we can cluster, create our UMAP and visualise. ``` r All.obj.sct <- FindNeighbors(All.obj.sct, reduction = "harmony", dims = 1:30) ``` ``` ## Computing nearest neighbor graph ``` ``` ## Computing SNN ``` ``` r All.obj.sct <- FindClusters(All.obj.sct, resolution = 0.3) ``` ``` ## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck ## ## Number of nodes: 8832 ## Number of edges: 287023 ## ## Running Louvain algorithm... ## Maximum modularity in 10 random starts: 0.9422 ## Number of communities: 14 ## Elapsed time: 0 seconds ``` ``` r All.obj.sct <- RunUMAP(All.obj.sct, dims = 1:30, reduction = "harmony") ``` ``` ## 11:35:34 UMAP embedding parameters a = 0.9922 b = 1.112 ``` ``` ## 11:35:34 Read 8832 rows and found 30 numeric columns ``` ``` ## 11:35:34 Using Annoy for neighbor search, n_neighbors = 30 ``` ``` ## 11:35:34 Building Annoy index with metric = cosine, n_trees = 50 ``` ``` ## 0% 10 20 30 40 50 60 70 80 90 100% ``` ``` ## [----|----|----|----|----|----|----|----|----|----| ``` ``` ## **************************************************| ## 11:35:36 Writing NN index file to temp file /var/folders/0_/8lnwn5sn7hn9nr8qv2jg48nw0000gn/T//RtmpUTDURW/file16eb27dfe7d ## 11:35:36 Searching Annoy index using 1 thread, search_k = 3000 ## 11:35:38 Annoy recall = 100% ## 11:35:39 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30 ## 11:35:41 Initializing from normalized Laplacian + noise (using RSpectra) ## 11:35:41 Commencing optimization for 500 epochs, with 364314 positive edges ## 11:35:54 Optimization finished ``` ``` r DimPlot(All.obj.sct, reduction = "umap", group.by = c("orig.ident", "SCT_snn_res.0.3")) ``` 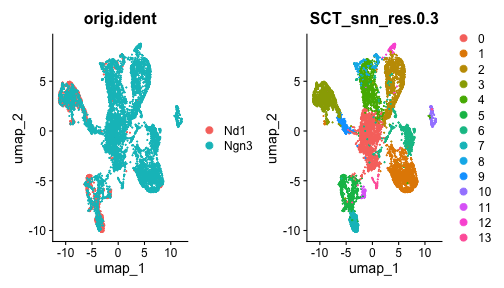<!-- --> --- ## scTransform. For downstream marker analysis it is recommended we normalise our data across batchs using the **PrepSCTFindMarkers** function. ``` r All.obj.sct <- PrepSCTFindMarkers(All.obj.sct) ``` ``` ## Found 2 SCT models. Recorrecting SCT counts using minimum median counts: 16788 ``` --- ## scTransform. Following this we can run FindMarkers on our data to identify markers across samples. ``` r harmonySCT.markers <- FindMarkers(All.obj.sct, ident.1 = 3, verbose = FALSE) VlnPlot(All.obj.sct, features = "Tph1", split.by = "orig.ident") ``` 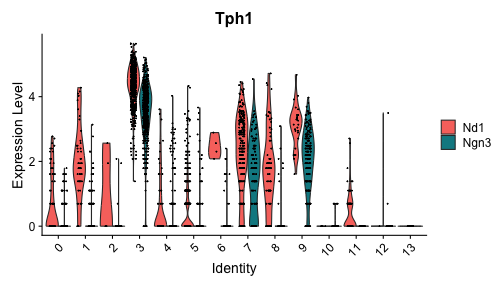<!-- -->