class: center, middle, inverse, title-slide # Visualizing Genomics Data (part 4) <html> <div style="float:left"> </div> <hr color='#EB811B' size=1px width=796px> </html> ### <a href="http://rockefelleruniversity.github.io/RU_VisualizingGenomicsData/" class="uri">http://rockefelleruniversity.github.io/RU_VisualizingGenomicsData/</a> --- ##The Course * Plotting Epigenomic data * Plotting Motifs occurrence --- ##Reminder of file types In this session we will be dealing with a range of data types. For more information on file types you can revisit our material. * [File Formats](https://rockefelleruniversity.github.io/Genomic_Data/). For more information on visualizing genomics data in browsers you can visit our IGV course. * [IGV](https://rockefelleruniversity.github.io/IGV_course/). --- ##Reminder of data types in Bioconductor We will also encounter and make use of many data structures and data types which we have seen throughout our courses on HTS data. You can revisit this material to refresh on HTS data analysis in Bioconductor and R below. * [Bioconductor](https://rockefelleruniversity.github.io/Bioconductor_Introduction/) * [Genomic Intervals](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/singlepage/GenomicIntervals_In_Bioconductor.html) * [Genomic Scores](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/singlepage/GenomicScores_In_Bioconductor.html) * [Sequences](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/singlepage/SequencesInBioconductor.html) * [Gene Models](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/singlepage/GenomicFeatures_In_Bioconductor.html) * [Alignments](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/singlepage/AlignedDataInBioconductor.html) * [ChIPseq](http://rockefelleruniversity.github.io/RU_ChIPseq/) * [ATAC-seq](http://rockefelleruniversity.github.io/RU_ATACseq/) * [RNA-seq](http://rockefelleruniversity.github.io/RU_RNAseq/) --- ##Materials All material for this course can be found on github. * [Visualizing Genomics Data](https://github.com/RockefellerUniversity/RU_VisualizingGenomicsData) Or can be downloaded as a zip archive from here. * [Download zip](https://github.com/RockefellerUniversity/RU_VisualizingGenomicsData/archive/master.zip) --- ##Materials Once the zip file in unarchived. All presentations as HTML slides and pages, their R code and HTML practical sheets will be available in the directories underneath. * **r_course/presentations/Slides/** Presentations as an HTML slide show. * **r_course/presentations/exercises/** Some tasks/examples to work through. --- ##Materials All data to run code in the presentations and in the practicals is available in the zip archive. This includes coverage as bigWig files, aligned reads as BAM files and genomic intervals stored as BED files. We also include some RData files containing precompiled results from querying database (in case of external server downtime). All data can be found under the **data** directory **data/** --- ##Set the Working directory Before running any of the code in the practicals or slides we need to set the working directory to the folder we unarchived. You may navigate to the unarchived visualizingGenomicsData folder in the Rstudio menu **Session -> Set Working Directory -> Choose Directory** or in the console. ```r setwd("/PathToMyDownload/VisualizingGenomicsData/r_course/") # e.g. setwd("~/Downloads/VisualizingGenomicsData/r_course") ``` --- ## Recap Previously we have reviewed how we can review signal and annotation over individual sites using and genome browser such as IGV and or programmatically using Gviz. .pull-left[  ] .pull-right[  ] --- ## Recap With many high throughput sequencing experiments we are able to gain a genome wide view of our assays (in RNA-seq we measure every gene's expression, in ChIPseq we identify all events for a user-defined transcription factor/mark, in ATAC-seq we evaluate all open/accessible regions in the genome.)  --- ## Recap Dimension reduction and clustering are very useful tools for simplifying the interpretation of this highly complex and dimensional biological data. .pull-left[ 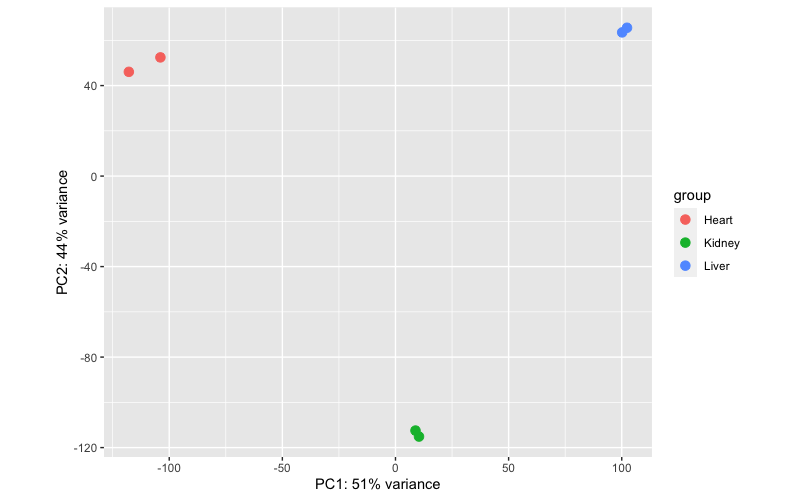 ] .pull-right[  ] --- ## Vizualizing Biological data As with RNAseq, epigenomics data can be displayed once reduced in dimension, such as with meta gene and profile plots, as well as with heatmaps. <div align="center"> <img src="imgs/large.png" alt="igv" height="400" width="800"> </div> --- ## Vizualizing Biological data In this session will review a few approaches to visualize epigenetics data. There are many alternate packages for visualizing epigenetic data in Bioconductor (soGGi,heatmaps and enrichmentmap packages) and in R (ngsPlot). Useful software outside R include Homer and Deeptools. --- ## CTCF ChIPseq In our ChIPseq session, we identified differential occupancy of Myc transcription factor between Mel and Ch12 cell lines. Here we will assess CTCF signal difference between Mel and Ch12. First lets read in some CTCF peaks into a GRangesList ```r peakFiles <- dir("data/CTCFpeaks/",pattern="*.peaks", full.names = TRUE) macsPeaks_GR <- list() for(i in 1:length(peakFiles)){ macsPeaks_DF <- read.delim(peakFiles[i], comment.char="#") macsPeaks_GR[[i]] <- GRanges( seqnames=macsPeaks_DF[,"chr"], IRanges(macsPeaks_DF[,"start"], macsPeaks_DF[,"end"] ) ) } macsPeaks_GRL <- GRangesList(macsPeaks_GR) names(macsPeaks_GRL) <- c("Ch12_1","Ch12_2","Mel_1","Mel_2") ``` --- ## CTCF ChIPseq Now we can produce our non-redundant set of peaks and score each of the peaks by their occurrence in our Mel and Ch12 samples. ```r allPeaksSet_nR <- reduce(unlist(macsPeaks_GRL)) overlap <- list() for(i in 1:length(macsPeaks_GRL)){ overlap[[i]] <- allPeaksSet_nR %over% macsPeaks_GRL[[i]] } overlapMatrix <- do.call(cbind,overlap) colnames(overlapMatrix) <- names(macsPeaks_GRL) mcols(allPeaksSet_nR) <- overlapMatrix allPeaksSet_nR[1,] ``` ``` ## GRanges object with 1 range and 4 metadata columns: ## seqnames ranges strand | Ch12_1 Ch12_2 Mel_1 Mel_2 ## <Rle> <IRanges> <Rle> | <logical> <logical> <logical> <logical> ## [1] chr1 3012656-3012773 * | TRUE TRUE FALSE FALSE ## ------- ## seqinfo: 21 sequences from an unspecified genome; no seqlengths ``` --- ## CTCF ChIPseq With the overlap matrix we can define our set of high-confidence peaks ```r HC_Peaks <- allPeaksSet_nR[ rowSums(as.data.frame( mcols(allPeaksSet_nR)[,c("Ch12_1","Ch12_2")])) >= 2 | rowSums(as.data.frame( mcols(allPeaksSet_nR)[,c("Mel_1","Mel_2")])) >= 2 ] HC_Peaks[1:2,] ``` ``` ## GRanges object with 2 ranges and 4 metadata columns: ## seqnames ranges strand | Ch12_1 Ch12_2 Mel_1 Mel_2 ## <Rle> <IRanges> <Rle> | <logical> <logical> <logical> <logical> ## [1] chr1 3012656-3012773 * | TRUE TRUE FALSE FALSE ## [2] chr1 3448274-3448415 * | FALSE FALSE TRUE TRUE ## ------- ## seqinfo: 21 sequences from an unspecified genome; no seqlengths ``` --- ## Defining a consensus peak set We can count the number of reads overlapping our peaks using the **summarizeOverlaps** function and a BamFileList of our BAMs to count. ```r library(Rsamtools) library(GenomicAlignments) bams <- c("~/Projects/Results/chipseq/testRun/BAMs/Sorted_CTCF_Ch12_1.bam", "~/Projects/Results/chipseq/testRun/BAMs/Sorted_CTCF_Ch12_2.bam", "~/Projects/Results/chipseq/testRun/BAMs/Sorted_CTCF_Mel_1.bam", "~/Projects/Results/chipseq/testRun/BAMs/Sorted_CTCF_Mel_2.bam") bamFL <- BamFileList(bams,yieldSize = 5000000) myCTCFCounts <- summarizeOverlaps(HC_Peaks, reads = bamFL, ignore.strand = TRUE) colData(myCTCFCounts)$Group <- c("Ch12","Ch12","Mel","Mel") ``` --- ## Differential ChIP Now we can use the DESeq2 package to compare our 2 cell-lines. ```r library(DESeq2) deseqCTCF <- DESeqDataSet(myCTCFCounts,design = ~ Group) deseqCTCF <- DESeq(deseqCTCF) CTCF_Ch12MinusMel <- results(deseqCTCF, contrast = c("CellLine","Mel","Ch12"), format="GRanges") ``` --- ## Generating rlog counts We can also use DESeq2 to give us some normalized, transformed data for visualization using **rlog**. ```r CTCFrlog <- rlog(deseqCTCF) CTCFrlog ``` --- ## Assessing the dataset With our newly created rlog transformed CTCF signal data we can create similar visualization as we have for RNAseq. First we can create our measures of sample distances. ```r sampleDists <- as.dist(1-cor(assay(CTCFrlog))) sampleDistMatrix <- as.matrix(sampleDists) pheatmap(sampleDistMatrix, clustering_distance_rows = sampleDists, clustering_distance_cols = sampleDists) ``` <!-- --> --- ## Assessing the dataset We can also review our dimension reduction by PCA to assess differences within and between groups with the **plotPCA**. Here we use by default the top 500 most variable sites. ```r plotPCA(CTCFrlog,intgroup="Group") ``` <!-- --> --- ## Assessing the dataset We can rerun using all sites to see some additional variance in PC2. ```r plotPCA(CTCFrlog,intgroup="Group", ntop=nrow(CTCFrlog)) ``` <!-- --> --- ## Assessing the dataset The plotPCA can return a data.frame of the PC scores for samples used in plot. ```r myData <- plotPCA(CTCFrlog,intgroup="Group", ntop=nrow(CTCFrlog), returnData=TRUE) myData ``` ``` ## PC1 PC2 group Group name ## CTCF_Ch12_1 -95.59844 17.12250 CTCF_Ch12 CTCF_Ch12 CTCF_Ch12_1 ## CTCF_Ch12_2 -91.08241 -17.63094 CTCF_Ch12 CTCF_Ch12 CTCF_Ch12_2 ## CTCF_MEL_1 94.06208 -11.13739 CTCF_Mel CTCF_Mel CTCF_MEL_1 ## CTCF_MEL_2 92.61876 11.64583 CTCF_Mel CTCF_Mel CTCF_MEL_2 ``` --- ## Assessing the dataset So we can use this data.frame and any additional information to produce a customized version of this plot. Here we add fragment length information. ```r myData$FragmentLength <- c(129,136,133,125) library(ggplot2) ggplot(myData,aes(x=PC1,y=PC2, colour=Group,size=FragmentLength))+ geom_point()+ scale_size_continuous(range=c(2,10)) ``` <!-- --> --- ## Assessing the dataset We can extract information on the influence of sites to the separation of samples along PC1 as we have with RNAseq. ```r pcRes <- prcomp(t(assay(CTCFrlog))) RankedPC1 <- rownames(pcRes$rotation)[order(pcRes$rotation[,1], decreasing=T)] RankedPC1[1:3] ``` ``` ## [1] "ID_chr4_89129278_89129824" "ID_chr4_89145405_89145765" ## [3] "ID_chrX_159987900_159988285" ``` --- ## Assessing the dataset We can see from the IGV that these top sites are Mel cell-line specific CTCF peaks.  --- ## Assessing the dataset We can also plot our significantly differential sites as defined by our DESeq2 test in a gene by sample heatmaps. ```r library(pheatmap) rlogMat <- assay(CTCFrlog) Diff <- names(CTCF_Ch12MinusMel)[CTCF_Ch12MinusMel$padj < 0.05 & !is.na(CTCF_Ch12MinusMel$padj) & abs(CTCF_Ch12MinusMel$log2FoldChange) > 3] sigMat <- rlogMat[rownames(rlogMat) %in% Diff,] pheatmap(sigMat,scale="row",show_rownames = FALSE) ``` <!-- --> --- ## soGGi The soGGi package contains a set of tools to profile high-throughput signalling data and motif occurrence over a set of genomic locations. We have used this earlier to produce some plots of our ATACseq signal split by fragment length over TSS regions and our cut-site footprints around CTCF motifs. .pull-left[  ] .pull-right[  ] --- ## soGGi We will use the development version of soGGi package [available from Github](https://github.com/ThomasCarroll/soGGi) to take advantage of the latest features. ```r library(devtools) install_github("ThomasCarroll/soGGi") ``` ```r library(soGGi) ``` --- ## soGGi Before we start plotting over regions we can update our DEseq2 results GRanges with some more information on the differential binding sites. This is some categorical data, based on the direction and significance of change. ```r UpInMel <- names(CTCF_Ch12MinusMel)[CTCF_Ch12MinusMel$padj < 0.05 & !is.na(CTCF_Ch12MinusMel$padj) & CTCF_Ch12MinusMel$log2FoldChange < -3] DownInMel <- names(CTCF_Ch12MinusMel)[CTCF_Ch12MinusMel$padj < 0.05 & !is.na(CTCF_Ch12MinusMel$padj) & abs(CTCF_Ch12MinusMel$log2FoldChange) > 3] SigVec <- ifelse(names(CTCF_Ch12MinusMel) %in% UpInMel,"UpInMel", ifelse(names(CTCF_Ch12MinusMel) %in% DownInMel,"DownInMel", "Other")) CTCF_Ch12MinusMel$Sig <- SigVec CTCF_Ch12MinusMel$name <- names(CTCF_Ch12MinusMel) ``` --- ## soGGi We also add some information on CTCF site signal strength. We are dividing our peaks into two groups: those that have high or low signal. ```r grPs <- quantile(CTCF_Ch12MinusMel$baseMean,c(0.33,0.66)) high <- names(CTCF_Ch12MinusMel)[CTCF_Ch12MinusMel$baseMean > grPs[2]] low <- names(CTCF_Ch12MinusMel)[CTCF_Ch12MinusMel$baseMean < grPs[2]] Strength <- ifelse(names(CTCF_Ch12MinusMel) %in% high,"high", ifelse(names(CTCF_Ch12MinusMel) %in% low,"low", "") ) CTCF_Ch12MinusMel$Strength <- Strength ``` --- ## soGGi Now we have our updated GRanges of DEseq2 results with our custom annotation. ```r CTCF_Ch12MinusMel[1:2,] ``` ``` ## GRanges object with 2 ranges and 9 metadata columns: ## seqnames ranges strand | baseMean ## <Rle> <IRanges> <Rle> | <numeric> ## ID_chr16_11670061_11670805 chr16 11670061-11670805 * | 3608.19 ## ID_chr1_75559493_75559979 chr1 75559493-75559979 * | 1400.68 ## log2FoldChange lfcSE stat pvalue ## <numeric> <numeric> <numeric> <numeric> ## ID_chr16_11670061_11670805 3.73522 0.121393 30.7696 6.69175e-208 ## ID_chr1_75559493_75559979 4.23851 0.139268 30.4341 1.94252e-203 ## padj Sig name ## <numeric> <character> <character> ## ID_chr16_11670061_11670805 4.12473e-203 DownInMel ID_chr16_11670061_11.. ## ID_chr1_75559493_75559979 5.98675e-199 DownInMel ID_chr1_75559493_755.. ## Strength ## <character> ## ID_chr16_11670061_11670805 high ## ID_chr1_75559493_75559979 high ## ------- ## seqinfo: 21 sequences from an unspecified genome; no seqlengths ``` --- ## soGGi We can use the **regionPlot()** function with our normalised bigWig of CTCF signal by setting **format** parameter to **bigwig**. Additionally we set the plot area width with the **distanceAround** parameter. ```r rownames(CTCF_Ch12MinusMel) <- NULL CTCFbigWig <- "data/Sorted_CTCF_Ch12_1Normalised.bw" CTCF_ch12 <- regionPlot(CTCFbigWig, testRanges = CTCF_Ch12MinusMel, style = "point", format = "bigwig", distanceAround = 750) ``` --- ## soGGi We can produce a simple profile plot by using the **plotRegion()** function with our **ChIPprofile** object. ```r p <- plotRegion(CTCF_ch12) p ``` <div align="center"> <img src="imgs/P8.png" alt="igv" height="350" width="600"> </div> --- ## soGGi As with our other packages, **soGGi** returns a **ggplot** object which can be further customized. ```r p+theme_bw()+ geom_point(colour="red") ``` <div align="center"> <img src="imgs/P9.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We can use the **summariseBy** argument to group our data for plotting by column information. Here by specify to group data by values in the **Strength** column. ```r plotRegion(CTCF_ch12, summariseBy = "Strength") ``` <div align="center"> <img src="imgs/P10.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We can supply additional groups to the **gts** argument as a list of sets of IDS/names to create groups from. By default the names are matched against the rownames of our **ChIPprofile** object. ```r rownames(CTCF_ch12) <- mcols(CTCF_ch12)$name plotRegion(CTCF_ch12, gts=list(high=high,low=low)) ``` <div align="center"> <img src="imgs/P11.png" alt="igv" height="300" width="600"> </div> --- ## soGGi When using the **gts** argument, we can supply the name of column to check group IDs/names against for grouping our plot to the **summariseBy** argument. ```r plotRegion(CTCF_ch12, gts=list(high=high,low=low), summariseBy = "name") ``` <div align="center"> <img src="imgs/P12.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We can also use lists of GRanges to group our data in soGGi. First we can create some TSS and TTS regions to investigate. ```r library(TxDb.Mmusculus.UCSC.mm10.knownGene) dsds <- genes(TxDb.Mmusculus.UCSC.mm10.knownGene) TSS <- resize(dsds,500,fix = "start") TTS <- resize(dsds,500,fix = "end") ``` --- ## soGGi We can then supply this list of GRanges to the **gts** parameter as we have our lists of IDs/names. Groups are defined by regions overlapping the specified ranges. ```r plotRegion(CTCF_ch12, gts=list(TSS=TSS, TTS=TTS)) ``` <div align="center"> <img src="imgs/P17.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We can control colouring with the **colourBy** argument. Here we specify **Group** to colour by our newly defined groups. ```r plotRegion(CTCF_ch12, summariseBy = "Strength", colourBy = "Group") ``` <div align="center"> <img src="imgs/P13.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We can control faceting with the **groupBy** argument. ```r plotRegion(CTCF_ch12, summariseBy = "Strength", colourBy = "Group", groupBy = "Group") ``` <div align="center"> <img src="imgs/P14.png" alt="igv" height="350" width="600"> </div> --- ## soGGi Again we can use ggplot basics to add some of our own colour schemes to our plots. ```r p <- plotRegion(CTCF_ch12, summariseBy = "Strength", colourBy = "Group", groupBy = "Group") cols <- c("high" = "darkred", "low" = "darkblue") p + scale_colour_manual(values = cols)+ theme_minimal() ``` <div align="center"> <img src="imgs/P15.png" alt="igv" height="200" width="400"> </div> --- ## soGGi We can use to soGGi to compare signal across samples. Here we load in some data now for CTCF signal in our Mel cell-line. ```r CTCFbigWig <- "data//Sorted_CTCF_MEL_1Normalised.bw" CTCF_mel <- regionPlot(CTCFbigWig, testRanges = CTCF_Ch12MinusMel, style = "point", format = "bigwig", distanceAround = 750) ``` --- ## soGGi We can combine **ChIPprofile** objects just as with vectors and lists using the **c()** function. ```r CTCF <- c(CTCF_mel,CTCF_ch12) CTCF ``` ``` ## class: ChIPprofile ## dim: 61639 1501 ## metadata(2): names AlignedReadsInBam ## assays(2): '' '' ## rownames(61639): giID2175 giID2176 ... giID59822 giID59823 ## rowData names(10): baseMean log2FoldChange ... Strength giID ## colnames(1501): Point_Centre-750 Point_Centre-749 ... Point_Centre749 ## Point_Centre750 ## colData names(0): ``` --- ## soGGi We can now plot our CTCF samples together in the same plot. We can control the plot by setting the **colourBy** argument to **Sample**. ```r plotRegion(CTCF, colourBy = "Sample") ``` <div align="center"> <img src="imgs/P20.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We could also use the sample to facet our plot. Here we set **groupBy** to **Sample**. ```r plotRegion(CTCF, groupBy = "Sample") ``` <div align="center"> <img src="imgs/P21.png" alt="igv" height="350" width="600"> </div> --- ## soGGi And we combine Sample and Grouping to produce some more complex plots. Here we group our data by the **Sig** column. ```r plotRegion(CTCF, summariseBy = "Sig", groupBy = "Group", colourBy="Sample") ``` <div align="center"> <img src="imgs/P22.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We have previously used **soGGi** to produce our plots of footprints across motifs. Now we will use motif occurrence as the signal to plot instead of regions to plot over. First we extract the CTCF motif and plot the seqLogo. ```r library(MotifDb) library(Biostrings) library(seqLogo) library(BSgenome.Mmusculus.UCSC.mm10) CTCFm <- query(MotifDb, c("CTCF")) ctcfMotif <- CTCFm[[1]] seqLogo(ctcfMotif) ``` <!-- --> --- ## soGGi We can use the **Biostrings** **matchPWM()** function to scan the genome for CTCF motifs, convert sites to a GRanges and this to an **rleList** using the coverage function. ```r myRes <- matchPWM(ctcfMotif, BSgenome.Mmusculus.UCSC.mm10[["chr19"]]) CTCFmotifs <- GRanges("chr19",ranges(myRes)) CTCFrle <- coverage(CTCFmotifs) CTCFrle[[1]] ``` ``` ## integer-Rle of length 61322860 with 5646 runs ## Lengths: 3055542 19 24798 19 ... 8069 19 13447 19 ## Values : 0 1 0 1 ... 0 1 0 1 ``` --- ## soGGi We can now plot our profile of motif occurrence across CTCF sites using the **regionPlot()** function and setting format parameter to **rlelist**. ```r motifPlot <- regionPlot(CTCFrle, CTCF_Ch12MinusMel, format = "rlelist") plotRegion(motifPlot) ``` <div align="center"> <img src="imgs/P25.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We can combine our ChIP profiles from CTCF signal in Mel and Motif occurrence and plot side by side. ```r CTCFSig19 <- CTCF_mel[seqnames(CTCF_mel) %in% "chr19",] rownames(motifPlot) <- rownames(CTCFSig19) <- NULL CTCFall <- c(motifPlot,CTCFSig19) p <- plotRegion(CTCFall) p ``` <div align="center"> <img src="imgs/P26.png" alt="igv" height="300" width="600"> </div> --- ## soGGi Since these two objects represent different data types, they will be in different scales. We can set the scales to resize within our facets by setting **freeScale** parameter to TRUE. ```r p <- plotRegion(CTCFall,freeScale=TRUE) p ``` <div align="center"> <img src="imgs/P27.png" alt="igv" height="300" width="600"> </div> --- ## soGGi In some instances we will want to plot over regions of unequal lengths, such as genes. ```r dsds <- genes(TxDb.Mmusculus.UCSC.mm10.knownGene) ``` ``` ## 66 genes were dropped because they have exons located on both strands ## of the same reference sequence or on more than one reference sequence, ## so cannot be represented by a single genomic range. ## Use 'single.strand.genes.only=FALSE' to get all the genes in a ## GRangesList object, or use suppressMessages() to suppress this message. ``` ```r names(dsds) <- NULL dsds ``` ``` ## GRanges object with 24528 ranges and 1 metadata column: ## seqnames ranges strand | gene_id ## <Rle> <IRanges> <Rle> | <character> ## [1] chr9 21062393-21073096 - | 100009600 ## [2] chr7 84935565-84964115 - | 100009609 ## [3] chr10 77711457-77712009 + | 100009614 ## [4] chr11 45808087-45841171 + | 100009664 ## [5] chr4 144157557-144162663 - | 100012 ## ... ... ... ... . ... ## [24524] chr3 84496093-85887516 - | 99889 ## [24525] chr3 110246109-110250998 - | 99890 ## [24526] chr3 151730922-151749960 - | 99899 ## [24527] chr3 65528410-65555518 + | 99929 ## [24528] chr4 136550540-136602723 - | 99982 ## ------- ## seqinfo: 66 sequences (1 circular) from mm10 genome ``` --- ## soGGi To produce average plots over genes we must rescale genes to be the same size.This is performed by splitting genes into equal number of bins, with bin size variable across genes and plotting as percent of regions. We can produce these plots in soGGi by setting **style** to **percentOfRegion**. ```r pol2Ser2 <- regionPlot(bamFile = "~/Downloads/ENCFF239XXP.bigWig", testRanges = dsds,style = "percentOfRegion", format = "bigwig") ``` --- ## soGGi Now we can plot the distribution of Polymerase 2 serine 2 across genes with **plotRegion()** function. ```r plotRegion(pol2Ser2) ``` <div align="center"> <img src="imgs/P30.png" alt="igv" height="350" width="600"> </div> --- ## soGGi We can now include the CTCF signal to review distibution of CTCF in genes compared to Pol2s2 signal. ```r CTCF_gene <- regionPlot(CTCFbigWig, testRanges = dsds, style = "percentOfRegion", format = "bigwig") ``` --- ## soGGi We can now plot the signal of CTCF and Pol2serine2 side by side. ```r rownames(CTCF_gene) <- rownames(pol2Ser2) <- NULL plotRegion(c(CTCF_gene,pol2Ser2), colourBy = "Sample", freeScale = TRUE, groupBy="Sample") ``` <div align="center"> <img src="imgs/P33.png" alt="igv" height="300" width="600"> </div> --- ## soGGi We may also want to review our CTCF profile data as heatmap of signal across our sites. We can produce heatmaps in soGGi using the **plotHeatmap** function. First we can extract a subset of sites to plot, here our sites which are higher in Mel. Since ChIPprofile objects act like GRanges we can subset by our metadata columns using mcols() function. ```r CTCF_Mel_Up <- CTCF[mcols(CTCF)$name %in% UpInMel,] ``` --- ## soGGi We can pass individual ChIPprofile objects to the plotHeatmap function to produce our heatmaps of CTCF signal in Mel sample. Here we set maxValue to make Heatmaps comparable across samples. ```r k1 <- plotHeatmap(CTCF_Mel_Up[[1]], col = colorRampPalette(blues9)(100), maxValue=15) ``` <div align="center"> <img src="imgs/P35.png" alt="igv" height="300" width="500"> </div> --- ## soGGi And we can produce the heatmap for our other sample in the same way. ```r k2 <- plotHeatmap(CTCF_Mel_Up[[2]], col = colorRampPalette(blues9)(100), maxValue=15) ``` <div align="center"> <img src="imgs/P36.png" alt="igv" height="300" width="500"> </div> --- ## profileplyr The heatmaps generated in soGGi however aren't as customizable as we would like. The **profileplyr** written by [Doug Barrows](https://dougbarrows.github.io) (Allis lab/BRC) provides a framework for plotting and manipulating genomic signal heatmaps. ```r library(profileplyr) ``` ``` ## ``` ``` ## Warning: replacing previous import 'ComplexHeatmap::pheatmap' by ## 'pheatmap::pheatmap' when loading 'profileplyr' ``` ``` ## ## Attaching package: 'profileplyr' ``` ``` ## The following object is masked from 'package:S4Vectors': ## ## params ``` --- ## profileplyr The profileplyr package has methods to import/export signal heatmaps from external sources such as DeepTools. Most usefully for us here though it can translate our soGGi ChIPprofile objects to profileplyr objects. The **profileplyr** object is a version of *Ranged*SummarizedExperiment object we should already recognize. ```r ctcfProPlyr <- as_profileplyr(CTCF) class(ctcfProPlyr) ``` ``` ## [1] "profileplyr" ## attr(,"package") ## [1] "profileplyr" ``` ```r ctcfProPlyr ``` ``` ## class: profileplyr ## dim: 61639 1501 ## metadata(0): ## assays(2): Sorted_CTCF_MEL_1Normalised.bw ## Sorted_CTCF_Ch12_1Normalised.bw ## rownames(61639): giID2175 giID2176 ... giID59822 giID59823 ## rowData names(12): baseMean log2FoldChange ... names sgGroup ## colnames(1501): Point_Centre-750 Point_Centre-749 ... Point_Centre749 ## Point_Centre750 ## colData names(0): ``` --- # profileplyr This means we can act on the object like any other *Ranged*SummarisedExperiment object including subsetting by GRanges. ```r ExampleGR <- GRanges("chr1:1-10000000") ctcfProPlyr[ctcfProPlyr %over% ExampleGR] ``` ``` ## class: profileplyr ## dim: 81 1501 ## metadata(0): ## assays(2): Sorted_CTCF_MEL_1Normalised.bw ## Sorted_CTCF_Ch12_1Normalised.bw ## rownames(81): giID2233 giID2238 ... giID6060 giID6128 ## rowData names(12): baseMean log2FoldChange ... names sgGroup ## colnames(1501): Point_Centre-750 Point_Centre-749 ... Point_Centre749 ## Point_Centre750 ## colData names(0): ``` --- # profileplyr Before we do any plotting we will subset our profileplyr object to just CTCF peaks changing between Mel and Ch12 cell-lines. We can use the mcols of our profileplyr object to subset to genes of interest. ```r ctcfProPlyr <- ctcfProPlyr[mcols(ctcfProPlyr)$name %in% c(UpInMel,DownInMel),] ``` --- # profileplyr Once we have the profileplyr object filtered to the data of interest, we can create our heatmap using the **generateEnrichedHeatmap** function. ```r generateEnrichedHeatmap(ctcfProPlyr) ``` <div align="center"> <img src="imgs/generateEnrichedHeatmap.png" alt="igv" height="300" width="400"> </div> --- # profileplyr We can perform clustering of our profileplyr object using the **clusterRanges()** function. By default this will perform hierarchical clustering and plot the result summarised within peaks. ```r ctcfProPlyr <- clusterRanges(ctcfProPlyr) ``` ``` ## No 'kmeans_k' or 'cutree_rows' arguments specified. profileplyr object will be returned new column with hierarchical order from hclust ``` <!-- --> --- # profileplyr We can now replot our heatmap using the new clustering. We must first tell the profileplyr object which metadata column to use for clustering/ordering the heatmap using the **orderBy** function. ```r ctcfProPlyr <- orderBy(tem,"hierarchical_order") generateEnrichedHeatmap(ctcfProPlyr) ``` <div align="center"> <img src="imgs/CTCF_ppClustered.png" alt="igv" height="300" width="400"> </div> --- ##Exercises Time for exercises! [Link here](../../exercises/exercises/Viz_part4_exercise.html) --- ##Solutions Time for solutions! [Link here](../../exercises/answers/Viz_part4_answers.html)