class: center, middle, inverse, title-slide .title[ # ChIPseq In Bioconductor (part1)<br /> <html><br /> <br /> <hr color='#EB811B' size=1px width=796px><br /> </html> ] .author[ ### Rockefeller University, Bioinformatics Resource Centre ] .date[ ### <a href="http://rockefelleruniversity.github.io/RU_ChIPseq/" class="uri">http://rockefelleruniversity.github.io/RU_ChIPseq/</a> ] --- ## ChIPseq Introduction Chromatin immunoprecipitation, followed by deep sequencing (**ChIPseq**) is a well established technique which allows for the genome wide identification of transcription factor binding sites and epigenetic marks. <div align="center"> <img src="imgs/chipOverview1.png" alt="igv" height="400" width="600"> </div> --- ## ChIPseq Introduction .pull-left[ <div align="center"> <img src="imgs/chipOverview2.png" alt="igv" height="500" width="300"> </div> ] .pull-right[ * Cross-linked and protein bound DNA. * Enrichment by antibody for specific protein or DNA state. * End repair, A-tailed and Illumina adapters added. * Fragments sequenced from either one/both ends. ] --- ## The data Our raw ChIPseq sequencing data will be in FASTQ format. <div align="center"> <img src="imgs/fq1.png" alt="igv" height="200" width="600"> </div> --- ## The data In this ChIPseq workshop we will be investigating the genome wide binding patterns of the transcription factor Myc in mouse MEL and Ch12 cell lines. We can retrieve the raw sequencing data from Encode website. Here we download the sequencing data for the Myc ChIPseq from the Mouse MEL cell line[, sample **ENCSR000EUA** (replicate 1), using the Encode portal.](https://www.encodeproject.org/experiments/ENCSR000EUA/) The direct link to the raw sequecing reads in FASTQ format can be found [here.](https://www.encodeproject.org/files/ENCFF001NQP/@@download/ENCFF001NQP.fastq.gz) Download the FASTQ for the other Myc MEL replicate from [sample ENCSR000EUA](https://www.encodeproject.org/experiments/ENCSR000EUA/). Direct link is [here](https://www.encodeproject.org/files/ENCFF001NQQ/@@download/ENCFF001NQQ.fastq.gz). --- class: inverse, center, middle # Working with raw ChIPseq data <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Working with raw ChIPseq data Once we have downloaded the raw FASTQ data we can use the [ShortRead package](https://bioconductor.org/packages/release/bioc/html/ShortRead.html) to review our sequence data quality. We have reviewed how to work with raw sequencing data in the [**FASTQ in Bioconductor** session.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#1) First we load the [ShortRead library.](https://bioconductor.org/packages/release/bioc/html/ShortRead.html) ``` r library(ShortRead) ``` --- ## Working with raw ChIPseq data First we will review the raw sequencing reads using functions in the [ShortRead package.](https://bioconductor.org/packages/release/bioc/html/ShortRead.html) This is similar to our QC we performed for RNAseq. We do not need to review all reads in the file to can gain an understanding of data quality. We can simply review a subsample of the reads and save ourselves some time and memory. Note when we subsample we retrieve random reads from across the entire FASTQ file. This is important as FASTQ files are often ordered by their position on the sequencer. --- ## Reading raw ChIPseq data We can subsample from a FASTQ file using functions in **ShortRead** package. Here we use the [**FastqSampler** and **yield** function](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#41) to randomly sample a defined number of reads from a FASTQ file. Here we subsample 1 million reads. This should be enough to have an understanding of the quality of the data. ``` r fqSample <- FastqSampler("~/Downloads/ENCFF001NQP.fastq.gz", n = 10^6) fastq <- yield(fqSample) ``` --- ## Working with raw ChIPseq data The resulting object is a [ShortReadQ object](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#10) showing information on the number of cycles, base pairs in reads, and number of reads in memory. ``` r fastq ``` ``` ## class: ShortReadQ ## length: 1000000 reads; width: 36 cycles ``` --- ## Raw ChIPseq data QC If we wished, we can assess information from the FASTQ file using our [familiar accessor functions.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#15) * **sread()** - Retrieve sequence of reads. * **quality()** - Retrieve quality of reads as ASCII scores. * **id()** - Retrieve IDs of reads. ``` r readSequences <- sread(fastq) readQuality <- quality(fastq) readIDs <- id(fastq) readSequences ``` ``` ## DNAStringSet object of length 1000000: ## width seq ## [1] 36 ATGAGAGTTCCTCTTTCTTACACATGTTTTTTTTTT ## [2] 36 GGTCANTGTGTTCAGTGTATGCTGCACTTACATTCC ## [3] 36 CTACCTGCTTCTTATCCAGCCCTCTCTTGTAATAGG ## [4] 36 GAATTGTTGATAATAACCTTATGCTTCTGTTGCTTA ## [5] 36 ATTCGTGGAGAGATAATGCGTGTATTTGGTTTTGTC ## ... ... ... ## [999996] 36 GAAATTCCAAAAACTATTTTTAGAACTTTACATATG ## [999997] 36 GTGGGGGCAGCAGACAAGTCCGGGGGAACAGTGAGC ## [999998] 36 CAAACAAACAAAACAAAACAAAACAAAAGAGAAGCA ## [999999] 36 TTGTATCCAGGAGAACCTTAGAATGTTCAGTGATGT ## [1000000] 36 AGGGACCGGCAAGTATTTCCCGCCTCATGTTTTGTC ``` --- ## Quality with raw ChIPseq data We can check some simple quality metrics for our subsampled FASTQ data. First, we can review the overall reads' quality scores. We use the [**alphabetScore()** function with our read's qualitys](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#28) to retrieve the sum quality for every read from our subsample. ``` r readQuality <- quality(fastq) readQualities <- alphabetScore(readQuality) readQualities[1:10] ``` ``` ## [1] 1109 1002 1190 1196 868 72 805 816 1041 1082 ``` --- ## Quality with raw ChIPseq data We can then produce a histogram of quality scores to get a better understanding of the distribution of scores. ``` r library(ggplot2) toPlot <- data.frame(ReadQ = readQualities) ggplot(toPlot, aes(x = ReadQ)) + geom_histogram() + theme_minimal() ``` ``` ## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`. ``` <!-- --> --- ## Base frequency with raw ChIPseq data We can review the occurrence of DNA bases within reads and well as the occurrence of DNA bases across sequencing cycles using the [**alphabetFrequency()**](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#18) and [**alphabetByCycle()**](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#30) functions respectively. Here we check the overall frequency of **A, G, C, T and N (unknown bases)** in our sequence reads. ``` r readSequences <- sread(fastq) readSequences_AlpFreq <- alphabetFrequency(readSequences) readSequences_AlpFreq[1:3, ] ``` ``` ## A C G T M R W S Y K V H D B N - + . ## [1,] 6 6 4 20 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ## [2,] 6 8 8 13 0 0 0 0 0 0 0 0 0 0 1 0 0 0 ## [3,] 6 12 5 13 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ``` --- ## Base frequency with raw ChIPseq data Once we have the frequency of DNA bases in our sequence reads we can retrieve the sum across all reads. ``` r summed__AlpFreq <- colSums(readSequences_AlpFreq) summed__AlpFreq[c("A", "C", "G", "T", "N")] ``` ``` ## A C G T N ## 10028851 7841813 7650350 10104255 374731 ``` --- ## Assess by cycle with raw ChIPseq data We can review DNA base occurrence by cycle using the [**alphabetByCycle()** function.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#30) ``` r readSequences_AlpbyCycle <- alphabetByCycle(readSequences) readSequences_AlpbyCycle[1:4, 1:10] ``` ``` ## cycle ## alphabet [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] ## A 307393 328036 282541 275320 276282 277553 278262 281389 271127 278521 ## C 179812 173882 205519 223468 210473 220596 213693 217991 220137 222141 ## G 242744 246446 227531 212323 212634 214609 210106 210020 213682 210753 ## T 265988 248240 279490 279258 290871 277974 288655 279444 284173 277758 ``` --- ## Assess by cycle with raw ChIPseq data We often plot this to visualise the base occurrence over cycles to observe any bias. First we arrange the base frequency into a data frame. ``` r AFreq <- readSequences_AlpbyCycle["A", ] CFreq <- readSequences_AlpbyCycle["C", ] GFreq <- readSequences_AlpbyCycle["G", ] TFreq <- readSequences_AlpbyCycle["T", ] toPlot <- data.frame(Count = c(AFreq, CFreq, GFreq, TFreq), Cycle = rep(1:36, 4), Base = rep(c("A", "C", "G", "T"), each = 36)) ``` --- ## Assess by cycle with raw ChIPseq data Now we can plot the frequencies using ggplot2 ``` r ggplot(toPlot, aes(y = Count, x = Cycle, colour = Base)) + geom_line() + theme_bw() ``` <!-- --> --- ## Assess by cycle with raw ChIPseq data We can also assess mean read quality over cycles. This will allow us to identify whether there are any isses with quality dropping off over time. For this we use the [**as(*read_quality*,"matrix")**](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#29) function first to translate our ASCII quality scores to numeric quality scores. ``` r qualAsMatrix <- as(readQuality, "matrix") qualAsMatrix[1:2, ] ``` ``` ## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12] [,13] [,14] [,15] [,16] ## [1,] 33 33 32 33 33 26 26 16 27 29 32 33 30 22 30 33 ## [2,] 33 33 18 25 22 4 20 33 31 33 30 33 32 28 32 31 ## [,17] [,18] [,19] [,20] [,21] [,22] [,23] [,24] [,25] [,26] [,27] [,28] [,29] [,30] ## [1,] 33 31 25 32 33 34 33 33 33 33 25 33 33 34 ## [2,] 33 24 33 31 33 27 30 31 27 19 25 33 30 30 ## [,31] [,32] [,33] [,34] [,35] [,36] ## [1,] 34 32 33 33 32 32 ## [2,] 28 24 30 29 25 22 ``` --- ## Assess by cycle with raw ChIPseq data We can now [visualise qualities across cycles using a boxplot.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/exercises/answers/fastq_answers.html) ``` r boxplot(qualAsMatrix[1:1000, ]) ``` <!-- --> --- ## Assess by cycle with raw ChIPseq data In this case the distribution of reads quality scores and read qualities over time look okay. We will often want to access FASTQ samples together to see if any samples stick out by these metrics. Here we observed a second population of low quality scores so will remove some reads with low quality scores and high unknown bases. --- class: inverse, center, middle # Filtering data <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Filtering data We will want to conserve our memory usage to [allow us to deal with loading large files.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#40) Here we set up a **FastqStreamer object** to read in 100000 reads at a time. ``` r fqStreamer <- FastqStreamer("~/Downloads/ENCFF001NQP.fastq.gz", n = 1e+05) ``` --- ## Filtering data Now we [loop through file](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#43), filter reads and [write out a FASTQ](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/FastQInBioconductor.html#39) of our filtered reads. We are filtering low quality reads and reads with many nonspecific (N) base calls. ``` r TotalReads <- 0 TotalReadsFilt <- 0 while (length(fq <- yield(fqStreamer)) > 0) { TotalReads <- TotalReads + length(fq) filt1 <- fq[alphabetScore(fq) > 300] filt2 <- filt1[alphabetFrequency(sread(filt1))[, "N"] < 10] TotalReadsFilt <- TotalReadsFilt + length(filt2) writeFastq(filt2, "filtered_ENCFF001NQP.fastq.gz", mode = "a") } ``` ``` r TotalReads ``` ``` ## [1] 25555179 ``` ``` r TotalReadsFilt ``` ``` ## [1] 22864597 ``` --- class: inverse, center, middle # Aligning data <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Aligning ChIPseq reads Following assessment of read quality and any read filtering we applied, we will want to align our reads to the genome so as to identify any genomic locations showing enrichment for aligned reads above background. Since ChIPseq reads will align continously against our reference genome we can use [our genomic aligners we have seen in previous sessions.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/AlignmentInBioconductor.html#7) The resulting BAM file will contain aligned sequence reads for use in further analysis. <div align="center"> <img src="imgs/sam2.png" alt="igv" height="200" width="600"> </div> --- ## Creating a reference genome First we need to retrieve the sequence information for the genome of interest in [FASTA format](https://rockefelleruniversity.github.io/Genomic_Data/presentations/slides/GenomicsData.html#9) We can use the [BSgenome libraries to retrieve the full sequence information.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/SequencesInBioconductor.html#4) For the mouse mm10 genome we load the package **BSgenome.Mmusculus.UCSC.mm10**. ``` r library(BSgenome.Mmusculus.UCSC.mm10) BSgenome.Mmusculus.UCSC.mm10 ``` ``` ## | BSgenome object for Mouse ## | - organism: Mus musculus ## | - provider: UCSC ## | - genome: mm10 ## | - release date: Sep 2017 ## | - 239 sequence(s): ## | chr1 chr2 chr3 chr4 ## | chr5 chr6 chr7 chr8 ## | chr9 chr10 chr11 chr12 ## | chr13 chr14 chr15 chr16 ## | chr17 chr18 chr19 chrX ## | ... ... ... ... ## | chrX_KQ030495_fix chrX_KQ030496_fix chrX_KQ030497_fix chrX_KZ289092_fix ## | chrX_KZ289093_fix chrX_KZ289094_fix chrX_KZ289095_fix chrY_JH792832_fix ## | chrY_JH792833_fix chrY_JH792834_fix chr1_KK082441_alt chr11_KZ289073_alt ## | chr11_KZ289074_alt chr11_KZ289075_alt chr11_KZ289077_alt chr11_KZ289078_alt ## | chr11_KZ289079_alt chr11_KZ289080_alt chr11_KZ289081_alt ## | ## | Tips: call 'seqnames()' on the object to get all the sequence names, call 'seqinfo()' to ## | get the full sequence info, use the '$' or '[[' operator to access a given sequence, see ## | '?BSgenome' for more information. ``` --- ## Creating a reference genome We will only use the major chromosomes for our analysis so we may exclude random and unplaced contigs. Here we cycle through the major chromosomes and create a [**DNAStringSet** object from the retrieved sequences](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/SequencesInBioconductor.html#17). ``` r mainChromosomes <- paste0("chr", c(1:19, "X", "Y", "M")) mainChrSeq <- lapply(mainChromosomes, function(x) BSgenome.Mmusculus.UCSC.mm10[[x]]) names(mainChrSeq) <- mainChromosomes mainChrSeqSet <- DNAStringSet(mainChrSeq) mainChrSeqSet ``` ``` ## DNAStringSet object of length 22: ## width seq names ## [1] 195471971 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chr1 ## [2] 182113224 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chr2 ## [3] 160039680 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chr3 ## [4] 156508116 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chr4 ## [5] 151834684 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chr5 ## ... ... ... ## [18] 90702639 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chr18 ## [19] 61431566 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chr19 ## [20] 171031299 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chrX ## [21] 91744698 NNNNNNNNNNNNNNNNNNNNNNNNNNN...NNNNNNNNNNNNNNNNNNNNNNNNNN chrY ## [22] 16299 GTTAATGTAGCTTAATAACAAAGCAAA...CTCTATTACGCAATAAACATTAACAA chrM ``` --- # Creating a reference genome. Now we have a **DNAStringSet** object we can use the [**writeXStringSet** to create our FASTA file of sequences to align to.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/SequencesInBioconductor.html#22) ``` r writeXStringSet(mainChrSeqSet, "BSgenome.Mmusculus.UCSC.mm10.mainChrs.fa") ``` --- ## Creating an Rsubread index We will be aligning using the **subjunc** algorithm from the folks behind subread. We can therefore use the **Rsubread** package. Before we attempt to align our FASTQ files, we will need to first build an index from our reference genome using the **buildindex()** function. The [**buildindex()** function simply takes the parameters of our desired index name and the FASTA file to build index from.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/AlignmentInBioconductor.html#14) REMEMBER: Building an index is memory intensive and by default is set to 8GB. This may be too large for your laptop or Desktop. Luckily we did this for RNAseq, so hopefully you will already have a built index. ``` r library(Rsubread) buildindex("mm10_mainchrs", "BSgenome.Mmusculus.UCSC.mm10.mainChrs.fa", memory = 8000, indexSplit = TRUE) ``` --- ## Rsubread ChIPseq alignment We can align our raw sequence data in FASTQ format to the new FASTA file of our mm10 genome sequence using the **Rsubread** package. Specifically we will be using the **align** function as this utilizes the subread genomic alignment algorithm. The [**align()** function accepts arguments for the index to align to, the FASTQ to align, the name of output BAM, the mode of alignment (rna or dna) and the phredOffset.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/AlignmentInBioconductor.html#15) Note that here we set the phredOffset to be 64. Rsubread may get upset if we set this wrong. ``` r myMapped <- align("mm10_mainchrs", "filtered_ENCFF001NQP.fastq.gz", output_format = "BAM", output_file = "Myc_Mel_1.bam", type = "dna", phredOffset = 64, nthreads = 4) ``` --- ## Rbowtie2 ChIPseq alignment One of the most well known group of alignment alogorthims are the Bowtie family. We can access Bowtie2 with the **Rbowtie2** package. The **QuasR** package allows access to the original Bowtie aligner, but it is a little slow and memory hungry. [Check out our session dedicated to alignment for using QuasR](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/AlignmentInBioconductor.html#36). ``` r library(Rbowtie2) ``` --- ## Rbowtie2 ChIPseq alignment As with **Rsubread**, the **Rbowtie2** package requires us to first to create an index to align to. We can do this using the **bowtie2_build()** function, specifying our FASTA file and desired name of index. ``` r bowtie2_build(references = "BSgenome.Mmusculus.UCSC.mm10.mainChrs.fa", bt2Index = file.path("BSgenome.Mmusculus.UCSC.mm10.mainChrs")) ``` --- ## Rbowtie2 ChIPseq alignment We can then align our FASTQ data using the **bowtie2()** function specifying our newly created index, the desired name of SAM output and an uncompressed FASTQ. We will need to uncompress our FASTQ then first. Here we use the **remove** is FALSE setting to maintain the original compressed FASTQ. ``` r library(R.utils) gunzip("filtered_ENCFF001NQP.fastq.gz", remove = FALSE) bowtie2(bt2Index = "BSgenome.Mmusculus.UCSC.mm10.mainChrs", samOutput = "ENCFF001NQP.sam", seq1 = "filtered_ENCFF001NQP.fastq") ``` --- # Rbowtie2 ChIPseq alignment Since Rbowtie2 also outputs a SAM file, we would need to need to convert to a BAM file. We can do this with the **RSamtools** **asBam()** function. ``` r bowtieBam <- asBam("ENCFF001NQP.sam") ``` --- ## Rbowtie2 ChIPseq alignment An important consideration when using Rbowtie2 is its input and output of uncompressed files. On the command line we may *stream* inputs to Rbowtie2, but in R this isnt an option (yet!) We would need to make sure we delete any temporary files created (SAM and/or uncompressed FASTQ) to avoid filling up our hard drives. We can delete files in R using the **unlink()** function. ``` r unlink("ENCFF001NQP.sam") ``` --- ## Sort and index reads As before, we sort and index our files using the [**Rsamtools** packages **sortBam()** and **indexBam()** functions respectively.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/AlignedDataInBioconductor.html#10) The resulting sorted and indexed BAM file is now ready for use in external programs such as IGV as well as for further downstream analysis in R. ``` r library(Rsamtools) sortBam("Myc_Mel_1.bam", "SR_Myc_Mel_rep1") indexBam("SR_Myc_Mel_rep1.bam") ``` --- class: inverse, center, middle # Mapped data <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Mapped reads Now we have the index for the BAM file, we can retrieve and plot the number of mapped reads using [the **idxstatsBam()** function.](https://rockefelleruniversity.github.io/Bioconductor_Introduction//presentations/slides/AlignedDataInBioconductor.html#16) ``` r mappedReads <- idxstatsBam("SR_Myc_Mel_rep1.bam") TotalMapped <- sum(mappedReads[, "mapped"]) ggplot(mappedReads, aes(x = seqnames, y = mapped)) + geom_bar(stat = "identity") + coord_flip() ``` <!-- --> --- ## Create a bigWig We can also create a bigWig from our sorted, indexed BAM file to allow us to quickly review our data in IGV. First we use the [**coverage()** function to create an **RLElist object** containing our coverage scores.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/Summarising_Scores_In_Bioconductor.html#13) ``` r forBigWig <- coverage("SR_Myc_Mel_rep1.bam") forBigWig ``` --- ## Create a bigWig We can now export our [**RLElist object** as a bigWig using the **rtracklayer** package's **export.bw()** function.](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/GenomicScores_In_Bioconductor.html#40) ``` r library(rtracklayer) export.bw(forBigWig, con = "SR_Myc_Mel_rep1.bw") ``` --- ## Create a bigWig We may wish to normalize our coverage to allow us to compare enrichment across samples. We can use the [**weight** parameter in the **coverage()**](https://rockefelleruniversity.github.io/Bioconductor_Introduction/presentations/slides/Summarising_Scores_In_Bioconductor.html#20) to scale our reads to the number of mapped reads multiplied by a million (reads per million). ``` r forBigWig <- coverage("SR_Myc_Mel_rep1.bam", weight = (10^6)/TotalMapped) forBigWig export.bw(forBigWig, con = "SR_Myc_Mel_rep1_weighted.bw") ``` --- # BAM and bigWig 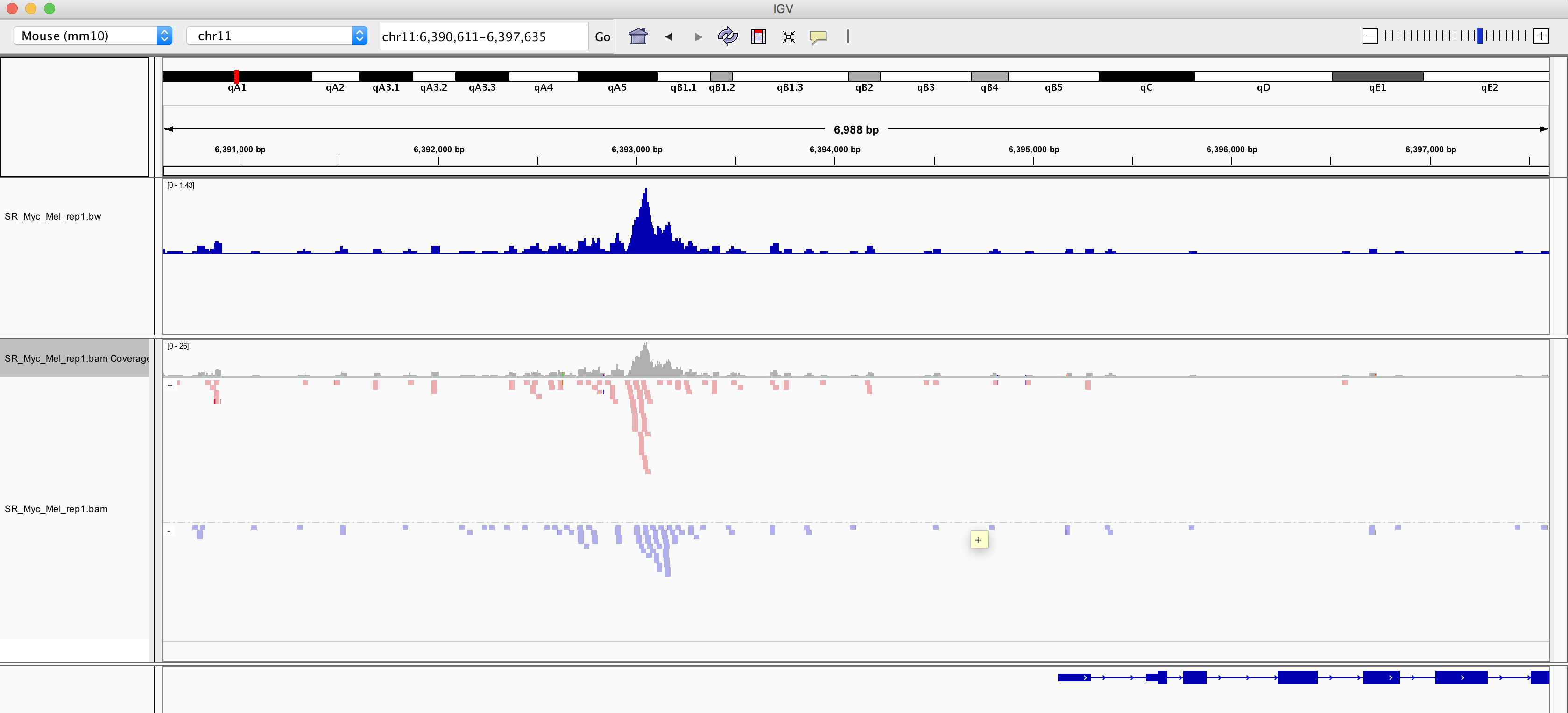 --- ## Time for an exercise! Exercise on ChIPseq data can be found [here](../../exercises/exercises/chipseq_part1_exercise.html) --- ## Answers to exercise Answers can be found [here](../../exercises/answers/chipseq_part1_answers.html) R code for solutions can be found [here](../../exercises/answers/chipseq_part1_answers.R)