class: center, middle, inverse, title-slide # ATACseq In Bioconductor <html> <div style="float:left"> </div> <hr color='#EB811B' size=1px width=796px> </html> ### Rockefeller University, Bioinformatics Resource Centre ### <a href="http://rockefelleruniversity.github.io/RU_ATACseq/" class="uri">http://rockefelleruniversity.github.io/RU_ATACseq/</a> --- ## Data In this session we will also keep working with the larger dataset from the ENCODE consortium generated by Bing Ren at UCSD. This dataset has multiple samples for multiple tissues (Liver, Kidney and Hindbrain), allowing us to perform a differential analysis on open regions. More details are [here](https://rockefelleruniversity.github.io/RU_ATACseq/presentations/slides/RU_ATAC_part2#5) --- ## ATACseq <div align="center"> <img src="imgs/mnATAC.jpg" alt="offset" height="300" width="600"> </div> * ATACseq - Uses transposases and offers a method to simultaneously extract signal from transcription factors binding sites and nucleosome positions from a single sample. --- ## ATACseq Unlike with ChIPseq where we know the target of IP, with ATACseq we know that a region is open/accessible but have no knowledge of what transcription factors may be present. We briefly saw at the end of our last session how we may scan for a specific motif under our peaks using **matchPWM**. In the upcoming sessions we will review how we can investigate motifs in ATACseq using motifmatchr. --- class: inverse, center, middle # Motif Databases <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Known Motif sources Bioconductor provides two major sources of motifs as database packages. These include: * The [MotifDb](https://www.bioconductor.org/packages/release/bioc/html/MotifDb.html) package. * The JASPAR databases, [JASPAR2020](https://www.bioconductor.org/packages/release/data/annotation/html/JASPAR2020.html) being latest. --- ## MotifDb The MotifDB package collects motif information from a wide range of sources and stores them in a DB object for use with other Bioconductor packages. ```r library(MotifDb) MotifDb ``` ``` ## MotifDb object of length 10701 ## | Created from downloaded public sources: 2013-Aug-30 ## | 10701 position frequency matrices from 21 sources: ## | cisbp_1.02: 874 ## | FlyFactorSurvey: 614 ## | HOCOMOCOv10: 1066 ## | HOCOMOCOv11-core-A: 181 ## | HOCOMOCOv11-core-B: 84 ## | HOCOMOCOv11-core-C: 135 ## | HOCOMOCOv11-secondary-A: 46 ## | HOCOMOCOv11-secondary-B: 19 ## | HOCOMOCOv11-secondary-C: 13 ## | HOCOMOCOv11-secondary-D: 290 ## | HOMER: 332 ## | hPDI: 437 ## | JASPAR_2014: 592 ## | JASPAR_CORE: 459 ## | jaspar2016: 1209 ## | jaspar2018: 1564 ## | jolma2013: 843 ## | ScerTF: 196 ## | stamlab: 683 ## | SwissRegulon: 684 ## | UniPROBE: 380 ## | 61 organism/s ## | Hsapiens: 5384 ## | Mmusculus: 1411 ## | Dmelanogaster: 1287 ## | Scerevisiae: 1051 ## | Athaliana: 803 ## | Celegans: 90 ## | other: 675 ## Scerevisiae-cisbp_1.02-M0001_1.02 ## Scerevisiae-cisbp_1.02-M0002_1.02 ## Scerevisiae-cisbp_1.02-M0003_1.02 ## Csativa-cisbp_1.02-M0004_1.02 ## Athaliana-cisbp_1.02-M0005_1.02 ## ... ## Mmusculus-UniPROBE-Zfp740.UP00022 ## Mmusculus-UniPROBE-Zic1.UP00102 ## Mmusculus-UniPROBE-Zic2.UP00057 ## Mmusculus-UniPROBE-Zic3.UP00006 ## Mmusculus-UniPROBE-Zscan4.UP00026 ``` --- ## MotifDb MotifDb object is special class of object called a **MotifList**. ```r class(MotifDb) ``` ``` ## [1] "MotifList" ## attr(,"package") ## [1] "MotifDb" ``` --- ## MotifDb Like standard List objects we can use length and names to get some information on our object ```r length(MotifDb) ``` ``` ## [1] 10701 ``` ```r MotifNames <- names(MotifDb) MotifNames[1:10] ``` ``` ## [1] "Scerevisiae-cisbp_1.02-M0001_1.02" "Scerevisiae-cisbp_1.02-M0002_1.02" ## [3] "Scerevisiae-cisbp_1.02-M0003_1.02" "Csativa-cisbp_1.02-M0004_1.02" ## [5] "Athaliana-cisbp_1.02-M0005_1.02" "Athaliana-cisbp_1.02-M0006_1.02" ## [7] "Athaliana-cisbp_1.02-M0007_1.02" "Athaliana-cisbp_1.02-M0008_1.02" ## [9] "Athaliana-cisbp_1.02-M0009_1.02" "Athaliana-cisbp_1.02-M0010_1.02" ``` --- ## Accesing MotifDb contents We can also access information directly from our list using standard list accessors. Here a **[** will subset to a single MotifList. Now we can see the information held in the MotifList a little more clearly. ```r MotifDb[1] ``` ``` ## MotifDb object of length 1 ## | Created from downloaded public sources: 2013-Aug-30 ## | 1 position frequency matrices from 1 source: ## | cisbp_1.02: 1 ## | 1 organism/s ## | Scerevisiae: 1 ## Scerevisiae-cisbp_1.02-M0001_1.02 ``` --- ## Accesing MotifDb contents A **[[** will subset to object within the element as with standard lists. Here we extract the position probability matrix. ```r MotifDb[[1]] ``` ``` ## 1 2 3 4 5 6 ## A 0.009615385 0.009615385 0.971153846 0.009615385 0.009615385 0.971153846 ## C 0.009615385 0.009615385 0.009615385 0.009615385 0.971153846 0.009615385 ## G 0.009615385 0.009615385 0.009615385 0.009615385 0.009615385 0.009615385 ## T 0.971153846 0.971153846 0.009615385 0.971153846 0.009615385 0.009615385 ## 7 8 ## A 0.009615385 0.009615385 ## C 0.971153846 0.009615385 ## G 0.009615385 0.009615385 ## T 0.009615385 0.971153846 ``` ```r colSums(MotifDb[[1]]) ``` ``` ## 1 2 3 4 5 6 7 8 ## 1 1 1 1 1 1 1 1 ``` --- ## Accesing MotifDb contents We can extract a DataFrame of all the motif metadata information using the **values()** function. ```r values(MotifDb)[1:2, ] ``` ``` ## DataFrame with 2 rows and 15 columns ## providerName providerId dataSource ## <character> <character> <character> ## Scerevisiae-cisbp_1.02-M0001_1.02 M0001_1.02 MA0000004_1.02 cisbp_1.02 ## Scerevisiae-cisbp_1.02-M0002_1.02 M0002_1.02 MA0000135_1.02 cisbp_1.02 ## geneSymbol geneId geneIdType ## <character> <character> <character> ## Scerevisiae-cisbp_1.02-M0001_1.02 ABF1 NA NA ## Scerevisiae-cisbp_1.02-M0002_1.02 AFT1 NA NA ## proteinId proteinIdType organism ## <character> <character> <character> ## Scerevisiae-cisbp_1.02-M0001_1.02 YKL112W SGD Scerevisiae ## Scerevisiae-cisbp_1.02-M0002_1.02 YGL071W SGD Scerevisiae ## sequenceCount bindingSequence bindingDomain ## <character> <character> <character> ## Scerevisiae-cisbp_1.02-M0001_1.02 NA NA NA ## Scerevisiae-cisbp_1.02-M0002_1.02 NA NA NA ## tfFamily experimentType pubmedID ## <character> <character> <character> ## Scerevisiae-cisbp_1.02-M0001_1.02 ABF1 NA 19111667 ## Scerevisiae-cisbp_1.02-M0002_1.02 AFT NA 19158363 ``` --- ## Accesing MotifDb contents We can use the **query** function to subset our MotifList by infomation in the metadata. ```r CTCFMotifs <- query(MotifDb, "CTCF") CTCFMotifs ``` ``` ## MotifDb object of length 19 ## | Created from downloaded public sources: 2013-Aug-30 ## | 19 position frequency matrices from 9 sources: ## | HOCOMOCOv10: 3 ## | HOCOMOCOv11-core-A: 2 ## | HOMER: 3 ## | JASPAR_2014: 2 ## | JASPAR_CORE: 1 ## | jaspar2016: 2 ## | jaspar2018: 3 ## | jolma2013: 1 ## | SwissRegulon: 2 ## | 4 organism/s ## | Hsapiens: 12 ## | Dmelanogaster: 3 ## | Mmusculus: 1 ## | other: 3 ## Hsapiens-HOCOMOCOv10-CTCFL_HUMAN.H10MO.A ## Hsapiens-HOCOMOCOv10-CTCF_HUMAN.H10MO.A ## Mmusculus-HOCOMOCOv10-CTCF_MOUSE.H10MO.A ## Hsapiens-HOCOMOCOv11-core-A-CTCFL_HUMAN.H11MO.0.A ## Hsapiens-HOCOMOCOv11-core-A-CTCF_HUMAN.H11MO.0.A ## ... ## Dmelanogaster-jaspar2018-CTCF-MA0531.1 ## Hsapiens-jaspar2018-CTCFL-MA1102.1 ## Hsapiens-jolma2013-CTCF ## Hsapiens-SwissRegulon-CTCFL.SwissRegulon ## Hsapiens-SwissRegulon-CTCF.SwissRegulon ``` --- ## Accesing MotifDb contents For more specific queries, multiple words can be used for filtering. ```r CTCFMotifs <- query(MotifDb, c("CTCF", "hsapiens", "jaspar2018")) CTCFMotifs ``` ``` ## MotifDb object of length 2 ## | Created from downloaded public sources: 2013-Aug-30 ## | 2 position frequency matrices from 1 source: ## | jaspar2018: 2 ## | 1 organism/s ## | Hsapiens: 2 ## Hsapiens-jaspar2018-CTCF-MA0139.1 ## Hsapiens-jaspar2018-CTCFL-MA1102.1 ``` --- ## JASPAR2020 package The JASPAR packages are updated more frequently and so may contain motifs not characterized in the MotifDb package. We can load the **JASPAR** package as usual and review the object as before. ```r library(JASPAR2020) JASPAR2020 ``` ``` ## An object of class "JASPAR2020" ## Slot "db": ## [1] "/usr/local/lib/R/host-site-library/JASPAR2020/extdata/JASPAR2020.sqlite" ``` --- ## JASPAR2020 and TFBSTools To interact with JASPAR package we will make use of the **TFBSTools** package from the same lab. Whereas the JASPAR package holds the information on Motifs and Position Probability Matrices (PPMs), TFBSTools has the functionality to manipulate and interact with these tools. Three useful functions available from TFBStools to interact with the JASPAR databases are the **getMatrixSet**, **getMatrixByID** and **getMatrixByID**. ```r library(TFBSTools) `?`(getMatrixByID) ``` --- ## TFBSTools for TFs The **getMatrixByID** and **getMatrixByName** take the JASPAR DB object and a JASPAR ID or transcription factor name respectively. Here we are using the transcription factor GATA2. The result is a new object class **PFMatrix**. ```r GATA2mat <- getMatrixByName(JASPAR2020, "GATA2") class(GATA2mat) ``` ``` ## [1] "PFMatrix" ## attr(,"package") ## [1] "TFBSTools" ``` --- ## TFBSTools for motifs JASPAR IDs are unique, so we can use them to select the exact motif we wanted. ```r GATA2mat <- getMatrixByID(JASPAR2020, "MA0036.2") ``` --- ## TFBSTools for motifs List accessors will not work here but we can retrieve names using the **ID** function. ```r ID(GATA2mat) ``` ``` ## [1] "MA0036.2" ``` --- ## Position Frequency Matrix To get hold of the Position Frequency Matrix (PFM) we can use the **Matrix** or **as.matrix** functions. ```r myMatrix <- Matrix(GATA2mat) myMatrixToo <- as.matrix(myMatrix) myMatrix ``` ``` ## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12] [,13] [,14] ## A 1569 1125 1555 1051 32 9 0 51 4380 0 0 1225 104 1256 ## C 567 768 729 525 1057 3015 192 0 0 0 4380 0 1765 1185 ## G 754 1709 1220 427 765 914 0 0 0 0 0 0 1967 224 ## T 1490 778 876 2377 2526 442 4188 4329 0 4380 0 3155 544 1715 ``` --- ## TFBSTools for motif sets We can also use the **getMatrixSet()** function to retrieve sets of motifs. We can specify a list of options for motifs we want to retrieve. To see the available filters use the help for getMatrixSet() function, ?getMatrixSet. ```r opts <- list() opts[["collection"]] <- "CORE" opts[["tax_group"]] <- "vertebrates" motifList <- getMatrixSet(JASPAR2020, opts) ``` --- class: inverse, center, middle # Visualizing motifs <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Visualizing motifs The seqLogo package offers a simple and intuitive way of visualizing our base frequencies within out motifs using the **seqLogo** function. A simple seqLogo shows the relative frequency of a base at each motif position by the relative size of the base compared to other bases at the same position. <!-- --> --- ## Visualizing motifs To create a seqLogo we must supply a PPM to the **seqLogo** function. Here we grab a PPM straight from MotidDb for CTCF. Here we set the **ic.scale** to FALSE to show the probability across the Y-axis ```r library(seqLogo) CTCFMotifs <- query(MotifDb, "CTCF") seqLogo::seqLogo(CTCFMotifs[[1]], ic.scale = FALSE) ``` <!-- --> --- ## Visualizing motifs It may be useful to plot the probabilities as information content. Information content here will range from 0 to 2 Bits. Positions with equal probabilities for each base will score 0 and positions with only 1 possible base will score 2. This allows you to quickly identify the important bases. ```r seqLogo::seqLogo(CTCFMotifs[[1]]) ``` 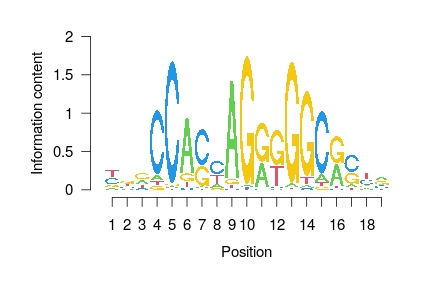<!-- --> --- ## Visualizing motifs The TFBSTools package and JASPAR provide us with point frequency matrices which we can't use directly in the seqLogo package ```r myMatrix ``` ``` ## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12] [,13] [,14] ## A 1569 1125 1555 1051 32 9 0 51 4380 0 0 1225 104 1256 ## C 567 768 729 525 1057 3015 192 0 0 0 4380 0 1765 1185 ## G 754 1709 1220 427 765 914 0 0 0 0 0 0 1967 224 ## T 1490 778 876 2377 2526 442 4188 4329 0 4380 0 3155 544 1715 ``` --- ## Visualizing motifs We can convert our point frequency matrix to a point probabilty matrix by simply dividing columns by their sum. ```r ppm <- myMatrix/colSums(myMatrix) ppm ``` ``` ## [,1] [,2] [,3] [,4] [,5] [,6] [,7] ## A 0.3582192 0.2568493 0.3550228 0.23995434 0.007305936 0.002054795 0.00000000 ## C 0.1294521 0.1753425 0.1664384 0.11986301 0.241324201 0.688356164 0.04383562 ## G 0.1721461 0.3901826 0.2785388 0.09748858 0.174657534 0.208675799 0.00000000 ## T 0.3401826 0.1776256 0.2000000 0.54269406 0.576712329 0.100913242 0.95616438 ## [,8] [,9] [,10] [,11] [,12] [,13] [,14] ## A 0.01164384 1 0 0 0.2796804 0.02374429 0.28675799 ## C 0.00000000 0 0 1 0.0000000 0.40296804 0.27054795 ## G 0.00000000 0 0 0 0.0000000 0.44908676 0.05114155 ## T 0.98835616 0 1 0 0.7203196 0.12420091 0.39155251 ``` --- ## Visualizing motifs We can then plot the result matrix using seqLogo. ```r seqLogo::seqLogo(ppm) ``` <!-- --> --- ## Visualizing motifs Fortunately, TFBSTools has its own version of seqLogo function we can use with one of its own **ICMatrix** classes. We simply need to convert our object with the **toICM()** function. ```r GATA2_IC <- toICM(GATA2mat) TFBSTools::seqLogo(GATA2_IC) ``` <!-- --> --- ## Visualizing with ggplot We can also use a ggplot2 style grammar of graphics with the **ggseqlogo** package. ```r library(ggseqlogo) library(ggplot2) ggseqlogo(myMatrix) + theme_minimal() ``` ``` ## Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = ## "none")` instead. ``` <!-- --> --- class: inverse, center, middle # Identifying Motifs in ATAC <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Known motif identification Since ATACseq only tells us which regions are open/accessible, we can use motifs within our ATACseq peak regions to identify potential transcription factors which may be acting there. The databases such as MotifDb and JASPAR2020, provide sets of known motif patterns for us to use our motif identification. --- ## motifmatchr To identify known motifs we will use the **motifmatchr** package which is a wrapper for the MOODS c++ library. This means that motifmatchr offers us a fast methods to identify motifs within our ATACseq data. We will use motifmatchr/MOODs with a default p-value cut-off. --- ## motifmatchr First we can retrieve a sensible set of motifs to scan for within our mouse tissue ATACseq data. Here we retrieve the vertebrate, JASPAR CORE motifs. We additional specify *all_versions* is FALSE to only include the latest version of a motif. ```r opts <- list() opts[["tax_group"]] <- "vertebrates" opts[["collection"]] <- "CORE" opts[["all_versions"]] <- FALSE motifsToScan <- getMatrixSet(JASPAR2020, opts) ``` --- ## motifmatchr The motifmatchr package main function is **matchMotifs()**. As with many Bioconductor functions , **matchMotifs** makes use of other Bioconductor objects such the BSGenome, GRanges and summarizedExperiment objects. We can review the full functionality by reading the help for matchMotifs, ?matchMotifs. --- ## ENCODE ATAC data We need some data to review so we can make use of the ENCODE mouse tissue ATAC data. We can load in the counts in a summarizedExperiment object from the **data** directory. ```r load("data/myCounts.RData") myCounts ``` ``` ## class: RangedSummarizedExperiment ## dim: 36320 6 ## metadata(0): ## assays(1): counts ## rownames: NULL ## rowData names(6): ## Sorted_Hindbrain_day_12_1_Small_Paired_peaks.narrowPeak ## Sorted_Hindbrain_day_12_2_Small_Paired_peaks.narrowPeak ... ## Sorted_Liver_day_12_1_Small_Paired_peaks.narrowPeak ## Sorted_Liver_day_12_2_Small_Paired_peaks.narrowPeak ## colnames(6): Sorted_Hindbrain_day_12_1_Small_Paired.bam ## Sorted_Hindbrain_day_12_2_Small_Paired.bam ... ## Sorted_Liver_day_12_1_Small_Paired.bam ## Sorted_Liver_day_12_2_Small_Paired.bam ## colData names(0): ``` --- ## ENCODE ATAC peaks We can retrieve the ranges of our peaks directly from our SummarizedExperiment object by using the standard accessor **rowRanges**. ```r peakRanges <- rowRanges(myCounts) peakRanges[1, ] ``` ``` ## GRanges object with 1 range and 6 metadata columns: ## seqnames ranges strand | ## <Rle> <IRanges> <Rle> | ## [1] chr1 3433954-3434095 * | ## Sorted_Hindbrain_day_12_1_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## Sorted_Hindbrain_day_12_2_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## Sorted_Kidney_day_15_1_Small_Paired_peaks.narrowPeak ## <logical> ## [1] TRUE ## Sorted_Kidney_day_15_2_Small_Paired_peaks.narrowPeak ## <logical> ## [1] TRUE ## Sorted_Liver_day_12_1_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## Sorted_Liver_day_12_2_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## ------- ## seqinfo: 22 sequences from an unspecified genome; no seqlengths ``` --- ## ENCODE ATAC peaks We can use the **getSeq()** function to extract sequences from a BSGenome object using a GRanges object to define the regions of interest. This is a similar approach to *de novo* motif discovery for [ChIPseq.](https://rockefelleruniversity.github.io/RU_ChIPseq/presentations/slides/ChIPseq_In_Bioconductor3.html#33) We load the BSgenome.Mmusculus.UCSC.mm10 library and re-center our peaks to 100bp using the resize function. Once resized we can extract the required sequences using the **getSeq()** function. ```r library(BSgenome.Mmusculus.UCSC.mm10) peakRangesCentered <- resize(peakRanges, fix = "center", width = 100) peakSeqs <- getSeq(BSgenome.Mmusculus.UCSC.mm10, peakRangesCentered) names(peakSeqs) <- as.character(peakRangesCentered) peakSeqs ``` ``` ## DNAStringSet object of length 36320: ## width seq names ## [1] 100 GGTTGTGCACTGTAAGCTGAC...CACTTCTGCTTCTGTCTGGCT chr1:3433975-3434074 ## [2] 100 AGTGCCTAGTCTTACTGCAAC...AGAAGAGGAGTAGGTGGGAGT chr1:3575937-3576036 ## [3] 100 CGCCGCCGCCGCCAGCAGCAG...GCGCGCTCCCTCCTCCCGCAG chr1:3671660-3671759 ## [4] 100 CTTCCCCCACCGCTGACACAC...TCTTCCCTTCACCCACCCTGA chr1:3994882-3994981 ## [5] 100 GAGGCAGCACCCTTTGTGGGC...GGCCTAAGCCACAGCAGCAGC chr1:4560256-4560355 ## ... ... ... ## [36316] 100 GCCATGTGCTCGCAGCTCCGC...CACGGCCCCGCCGGCTCCCCT chrY:90784203-907... ## [36317] 100 GGAGTTGTAGTCCTGCCGTAG...TGGGAGTTGCAGCGCCATGTT chrY:90784972-907... ## [36318] 100 CTCTGGAGACCAGGCTGACCT...ACCACGCCCGGATTTTTACCA chrY:90801528-908... ## [36319] 100 CTTCTCTGCCTCATGGGGACG...CGTCCCGGGCAGGCGGCGTCT chrY:90807427-908... ## [36320] 100 GGGGGGTTAGGTTAGGGTTAG...GGGGGTTAGGTTAGGGTTAGG chrY:90813049-908... ``` --- ## Finding motif positions As we saw in its help, The matchMotifs function can provide output of motif matches as matches, scores or positions. Here we scan for 4 selected motifs from JASPAR2020 using the sequence under the first 100 ATACseq peaks and specify the out as **positions**. ```r motif_positions <- matchMotifs(motifsToScan[1:4], peakSeqs[1:100], out = "positions") class(motif_positions) ``` ``` ## [1] "list" ``` ```r length(motif_positions) ``` ``` ## [1] 4 ``` --- ## Finding motif positions The result contains a list of the same length as the number of motifs tested. Each element contains a IRangeslist with an entry for every sequence tested and a IRanges of motif positions within the peak sequences. ```r motif_positions$MA0029.1 ``` ``` ## IRangesList object of length 100: ## [[1]] ## IRanges object with 0 ranges and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## ## [[2]] ## IRanges object with 0 ranges and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## ## [[3]] ## IRanges object with 0 ranges and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## ## ... ## <97 more elements> ``` --- ## Finding motif positions We can unlist our IRangeslist to a standard list for easier working. ```r MA0029hits <- motif_positions$MA0029.1 names(MA0029hits) <- names(peakSeqs[1:100]) unlist(MA0029hits, use.names = TRUE) ``` ``` ## IRanges object with 1 range and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## chr1:13566347-13566446 56 69 14 | + 10.0096 ``` --- ## Finding motif hits We may simply want to map motifs to their ATACseq peaks. To do this we can set the **out** parameter to **matches**. This will return a SummarizedExperiment object. ```r motifHits <- matchMotifs(motifsToScan, peakSeqs, out = "matches") class(motifHits) ``` ``` ## [1] "SummarizedExperiment" ## attr(,"package") ## [1] "SummarizedExperiment" ``` ```r motifHits ``` ``` ## class: SummarizedExperiment ## dim: 36320 746 ## metadata(0): ## assays(1): motifMatches ## rownames: NULL ## rowData names(0): ## colnames(746): MA0004.1 MA0006.1 ... MA0528.2 MA0609.2 ## colData names(1): name ``` --- ## Finding motif hits We can retrieve a matrix of matches by motif and peak using the motifMatches function. ```r mmMatrix <- motifMatches(motifHits) dim(mmMatrix) ``` ``` ## [1] 36320 746 ``` ```r mmMatrix[1:8, 1:8] ``` ``` ## 8 x 8 sparse Matrix of class "lgCMatrix" ## MA0004.1 MA0006.1 MA0019.1 MA0029.1 MA0030.1 MA0031.1 MA0040.1 MA0041.1 ## [1,] . . . . . . . . ## [2,] . . . . . . . . ## [3,] . . . . . . . . ## [4,] . . . . . . . . ## [5,] . . . . . . . . ## [6,] . . . . . . . . ## [7,] . | . . . . . . ## [8,] . . . . . . . . ``` --- ## Finding motif hits Although a sparse matrix, we can still use our matrix operations to extract useful information from this object. We can use the **colSums()** to identify the total occurrence of motifs in our peak sequences. ```r totalMotifOccurence <- colSums(mmMatrix) totalMotifOccurence[1:4] ``` ``` ## MA0004.1 MA0006.1 MA0019.1 MA0029.1 ## 1137 1303 184 1163 ``` --- ## Finding motif hits We can also identify peaks which contain a hit for a selected motif. ```r peaksWithMA0912 <- peakRangesCentered[mmMatrix[, "MA0912.2"] == 1] peaksWithMA0912 ``` ``` ## GRanges object with 361 ranges and 6 metadata columns: ## seqnames ranges strand | ## <Rle> <IRanges> <Rle> | ## [1] chr1 5232911-5233010 * | ## [2] chr1 9783650-9783749 * | ## [3] chr1 15644761-15644860 * | ## [4] chr1 31233772-31233871 * | ## [5] chr1 39393610-39393709 * | ## ... ... ... ... . ## [357] chrX 101332594-101332693 * | ## [358] chrX 101429501-101429600 * | ## [359] chrX 109012641-109012740 * | ## [360] chrX 166385018-166385117 * | ## [361] chrX 166672780-166672879 * | ## Sorted_Hindbrain_day_12_1_Small_Paired_peaks.narrowPeak ## <logical> ## [1] TRUE ## [2] FALSE ## [3] FALSE ## [4] TRUE ## [5] FALSE ## ... ... ## [357] TRUE ## [358] TRUE ## [359] FALSE ## [360] FALSE ## [361] FALSE ## Sorted_Hindbrain_day_12_2_Small_Paired_peaks.narrowPeak ## <logical> ## [1] TRUE ## [2] FALSE ## [3] FALSE ## [4] TRUE ## [5] FALSE ## ... ... ## [357] TRUE ## [358] TRUE ## [359] FALSE ## [360] FALSE ## [361] FALSE ## Sorted_Kidney_day_15_1_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## [2] FALSE ## [3] TRUE ## [4] FALSE ## [5] FALSE ## ... ... ## [357] TRUE ## [358] TRUE ## [359] FALSE ## [360] TRUE ## [361] FALSE ## Sorted_Kidney_day_15_2_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## [2] FALSE ## [3] TRUE ## [4] FALSE ## [5] FALSE ## ... ... ## [357] TRUE ## [358] TRUE ## [359] FALSE ## [360] TRUE ## [361] FALSE ## Sorted_Liver_day_12_1_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## [2] TRUE ## [3] FALSE ## [4] FALSE ## [5] TRUE ## ... ... ## [357] TRUE ## [358] TRUE ## [359] TRUE ## [360] FALSE ## [361] TRUE ## Sorted_Liver_day_12_2_Small_Paired_peaks.narrowPeak ## <logical> ## [1] FALSE ## [2] TRUE ## [3] FALSE ## [4] FALSE ## [5] TRUE ## ... ... ## [357] TRUE ## [358] TRUE ## [359] TRUE ## [360] FALSE ## [361] TRUE ## ------- ## seqinfo: 22 sequences from an unspecified genome; no seqlengths ``` --- class: inverse, center, middle # summarizing ATAC signal to Motifs <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## summarizing ATAC signal to Motifs The **chromVar** package allows for the summarization of ATACseq signal changes to the motifs within peaks. With this summarization we can potentially identify which motifs may have an important role in a set of ATAC samples compared to other samples. The chromVar package comes from the same lab and author as the motifmatchr package and so will work well together. First we load the library. ```r library(chromVAR) ``` --- ## Setting up for chromVar To identify motifs for chomVar we will use motifMatchr with a different set of inputs. Here we will provide our __*Ranged*SummarizedExperiment__ object contaitng our counts over peaks directly to the matchMotifs function. First we will remove any peaks with less than 5 reads across all samples. ```r myCounts <- myCounts[rowSums(assay(myCounts)) > 5, ] ``` --- ## Correct GC bias Next we can correct for any potential GC bias which may have arisen in sequencing. Previously we have seen that we have GC bias observable in our FastQ quality checks. For comparing across differing sets of peaks with differing sequences compositions we will need to correct for this bias. We can use the **addGCBias** and specify our genome as a BSgenome object to correct in chromVar. ```r myCounts <- addGCBias(myCounts, genome = BSgenome.Mmusculus.UCSC.mm10) ``` --- ## ID motifs in peaks Having corrected for bias, we can use the matchMotifs function again to identify motifs under our ATACseq peaks. Here we supply our __*Ranged*SummarizedExperiment__ of counts in peaks and the genome of interest to the **matchMotifs** function and use the default **out** of *matches*. ```r motif_ix <- matchMotifs(motifsToScan, myCounts, genome = BSgenome.Mmusculus.UCSC.mm10) motif_ix ``` ``` ## class: RangedSummarizedExperiment ## dim: 36320 746 ## metadata(0): ## assays(1): motifMatches ## rownames: NULL ## rowData names(7): ## Sorted_Hindbrain_day_12_1_Small_Paired_peaks.narrowPeak ## Sorted_Hindbrain_day_12_2_Small_Paired_peaks.narrowPeak ... ## Sorted_Liver_day_12_2_Small_Paired_peaks.narrowPeak bias ## colnames(746): MA0004.1 MA0006.1 ... MA0528.2 MA0609.2 ## colData names(1): name ``` --- ## Run chromVar Following the identification of motifs in our peaks, we can perform the summarization of ATACseq signal to motifs using the **computeDeviations** and the **computeVariability** functions. ```r deviations <- computeDeviations(object = myCounts, annotations = motif_ix) variability_Known <- computeVariability(deviations) ``` --- ## chromVar deviations The deviations result contains a SummarizedExperiment object with the Z-scores showing the enrichment for ATACseq signal in each sample for every motif. ```r devZscores <- deviationScores(deviations) devZscores[1:2, ] ``` ``` ## Sorted_Hindbrain_day_12_1_Small_Paired.bam ## MA0004.1 3.679156 ## MA0006.1 8.046464 ## Sorted_Hindbrain_day_12_2_Small_Paired.bam ## MA0004.1 5.431137 ## MA0006.1 7.633895 ## Sorted_Kidney_day_15_1_Small_Paired.bam ## MA0004.1 4.114432 ## MA0006.1 6.570759 ## Sorted_Kidney_day_15_2_Small_Paired.bam ## MA0004.1 4.564610 ## MA0006.1 6.004668 ## Sorted_Liver_day_12_1_Small_Paired.bam ## MA0004.1 -4.276874 ## MA0006.1 -8.348624 ## Sorted_Liver_day_12_2_Small_Paired.bam ## MA0004.1 -6.653514 ## MA0006.1 -6.824266 ``` --- ## chromVar variability The variability result contains the ranking of motifs by their variability across samples. Highly variable motifs may indicate motifs associated with a particular sample group or variable across all groups. ```r variability_Known <- variability_Known[order(variability_Known$p_value), ] variability_Known[1:10, ] ``` ``` ## name variability bootstrap_lower_bound bootstrap_upper_bound ## MA0140.2 GATA1::TAL1 31.47098 3.1948304 34.67008 ## MA0600.2 RFX2 17.77072 7.3944769 21.22610 ## MA0036.3 GATA2 32.01213 0.3921849 34.17913 ## MA0037.3 GATA3 25.31902 0.8039623 27.44567 ## MA0766.2 GATA5 25.01678 0.4178051 26.75562 ## MA0035.4 GATA1 26.04815 0.8122387 27.98797 ## MA0482.2 GATA4 27.45124 0.8642210 29.45822 ## MA1104.2 GATA6 29.73974 0.5806377 31.84163 ## MA0509.2 RFX1 17.52837 8.5454333 21.34927 ## MA0867.2 SOX4 16.47110 5.8375559 19.59171 ## p_value p_value_adj ## MA0140.2 0.000000e+00 0.000000e+00 ## MA0600.2 0.000000e+00 0.000000e+00 ## MA0036.3 0.000000e+00 0.000000e+00 ## MA0037.3 0.000000e+00 0.000000e+00 ## MA0766.2 0.000000e+00 0.000000e+00 ## MA0035.4 0.000000e+00 0.000000e+00 ## MA0482.2 0.000000e+00 0.000000e+00 ## MA1104.2 0.000000e+00 0.000000e+00 ## MA0509.2 0.000000e+00 0.000000e+00 ## MA0867.2 3.690799e-291 2.749645e-289 ``` --- ## chromVar results We can use the results from the variability with our Z-score deviations to identify in which sample our motifs are enriched. ```r topVariable <- variability_Known[1:20, ] devTop <- merge(topVariable[, 1, drop = FALSE], devZscores, by = 0) devTop[1:2, ] ``` ``` ## Row.names name Sorted_Hindbrain_day_12_1_Small_Paired.bam ## 1 MA0035.4 GATA1 -24.29333 ## 2 MA0036.3 GATA2 -29.55701 ## Sorted_Hindbrain_day_12_2_Small_Paired.bam ## 1 -22.56358 ## 2 -30.63070 ## Sorted_Kidney_day_15_1_Small_Paired.bam ## 1 -25.01967 ## 2 -30.24737 ## Sorted_Kidney_day_15_2_Small_Paired.bam ## 1 -24.19630 ## 2 -29.73203 ## Sorted_Liver_day_12_1_Small_Paired.bam Sorted_Liver_day_12_2_Small_Paired.bam ## 1 25.81764 26.97208 ## 2 31.46003 32.42450 ``` --- ## chromVar results A useful way to visualize these results is using a heatmap. Although we cover this in a later session, here we can use the **pheatmap** library with default settings to illustrate where our most variable motifs are active. ```r devToPlot <- as.matrix(devTop[, -c(1:2)]) rownames(devToPlot) <- devTop[, 2] library(pheatmap) pheatmap(devToPlot) ``` <!-- --> --- class: inverse, center, middle # Identifying Motifs *de novo* in ATAC <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## *De novo* motif discovery So far we have reviewed how to identify known motifs within our ATACseq peaks. In our [ChIPseq training](https://rockefelleruniversity.github.io/RU_ChIPseq/presentations/slides/ChIPseq_In_Bioconductor3.html#28) we cover howt we can use the Meme-ChIP software to perform *De novo* motif identification. Here we wll use the Meme-ChIP software in a discriminative mode to identify *De novo* motifs enriched in one set of samples over another. --- ## Differential peaks First we will need to identify a set of peaks enriched for ATACseq signal in one group over another. To do this we will use DESeq2 to identify ATACseq peaks enriched in 1 set over another. We can load our Liver, Kidney and Brain counts from earlier into a DEseq object for differential analysis. ```r require(DESeq2) load("data/myCounts.RData") Group <- factor(c("HindBrain", "HindBrain", "Kidney", "Kidney", "Liver", "Liver")) colData(myCounts) <- DataFrame(data.frame(Group, row.names = colnames(myCounts))) dds <- DESeqDataSet(myCounts, design = ~Group) dds <- DESeq(dds) ``` ``` ## estimating size factors ``` ``` ## estimating dispersions ``` ``` ## gene-wise dispersion estimates ``` ``` ## mean-dispersion relationship ``` ``` ## -- note: fitType='parametric', but the dispersion trend was not well captured by the ## function: y = a/x + b, and a local regression fit was automatically substituted. ## specify fitType='local' or 'mean' to avoid this message next time. ``` ``` ## final dispersion estimates ``` ``` ## fitting model and testing ``` --- ## Differential peaks We can then use the DESeq2 function **results** to identify differential ATAC regions and set the **format** parameter to **GRanges** to allow the return of the result as a GRanges object. ```r myRes <- results(dds, contrast = c("Group", "Liver", "Kidney"), format = "GRanges") myRes <- myRes[order(myRes$padj), ] upRegions <- myRes[myRes$log2FoldChange > 0][1:1000] downRegions <- myRes[myRes$log2FoldChange < 0, ][1:1000] upRegions ``` ``` ## GRanges object with 1000 ranges and 6 metadata columns: ## seqnames ranges strand | baseMean log2FoldChange ## <Rle> <IRanges> <Rle> | <numeric> <numeric> ## [1] chr7 100467072-100467556 * | 101.6408 3.99023 ## [2] chr18 32542348-32542812 * | 96.8956 4.11485 ## [3] chr10 30763197-30763615 * | 94.4364 3.78759 ## [4] chr15 103258321-103258745 * | 99.6945 3.59581 ## [5] chr8 123978898-123979311 * | 84.3669 4.13890 ## ... ... ... ... . ... ... ## [996] chr5 148897184-148897609 * | 24.8790 3.10487 ## [997] chr5 147888548-147888858 * | 21.1643 3.45870 ## [998] chr7 131425006-131425298 * | 25.7261 3.01058 ## [999] chr3 14886438-14886963 * | 63.2840 1.59576 ## [1000] chr8 122719754-122720057 * | 26.7802 2.94430 ## lfcSE stat pvalue padj ## <numeric> <numeric> <numeric> <numeric> ## [1] 0.290132 13.7532 4.87419e-43 1.77031e-38 ## [2] 0.304919 13.4949 1.67615e-41 3.04389e-37 ## [3] 0.281546 13.4528 2.96263e-41 3.58676e-37 ## [4] 0.281400 12.7783 2.16807e-37 1.96861e-33 ## [5] 0.330165 12.5359 4.75193e-36 3.45180e-32 ## ... ... ... ... ... ## [996] 0.487194 6.37297 1.85407e-10 5.79017e-09 ## [997] 0.543067 6.36883 1.90469e-10 5.94317e-09 ## [998] 0.472801 6.36754 1.92089e-10 5.98855e-09 ## [999] 0.250669 6.36602 1.93994e-10 6.04275e-09 ## [1000] 0.462528 6.36567 1.94435e-10 6.05130e-09 ## ------- ## seqinfo: 22 sequences from an unspecified genome; no seqlengths ``` --- ## Resize peaks For Meme-ChIP we will want to resize the regions to 100bp. By default Meme-ChIP will perform this trimming for us but to ensure it is in the centre we will do this upfront. ```r upRegions <- resize(upRegions, fix = "center", width = 100) downRegions <- resize(downRegions, fix = "center", width = 100) ``` --- ## Peak sequences We can now use the **getSeq** function as previously to extract the signal from around within the GRange regions and write to a FASTA file for use in Meme-ChIP using the **writeXStringSet** function. ```r library(BSgenome.Mmusculus.UCSC.mm10) upStrings <- getSeq(BSgenome.Mmusculus.UCSC.mm10, upRegions) downStrings <- getSeq(BSgenome.Mmusculus.UCSC.mm10, downRegions) names(upStrings) <- as.character(upRegions) names(downStrings) <- as.character(downRegions) writeXStringSet(upStrings, file = "UpRegions.fa") writeXStringSet(downStrings, file = "DownStrings.fa") ``` --- ## Meme-ChIP We can then submit the sample to the Meme-ChIP online submission form found [here](http://meme-suite.org/tools/meme-chip). In contrast to for ChIPseq, here we will run this in Differential mode to compare our sequences in Liver enriched regions to those depleted in Liver. The results can be found in **data/memeResult_LiverVsBrain** --- ## Using *De novo* motifs We may want to use the motifs discovered by Meme-ChIP in other Bioconductor software. To do this we can use the **universalmotif** package. ```r library(universalmotif) ``` --- ## Importing motifs The **universalmotif** package provides lots of useful functions for importing motif sets. One useful and relevant function is the **read_meme()** function. ```r memeMotifs <- read_meme("data/memeResult_LiverVsBrain/combined.meme") memeMotifs ``` ``` ## [[1]] ## ## Motif name: 1 ## Alternate name: HGATA-DREME-1 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 8.78 ## Pseudocount: 1 ## Consensus: WGATA ## Target sites: 1236 ## E-value: 7.6e-275 ## Extra info: [eval.string] 7.6e-275 ## ## W G A T A ## A 0.58 0 1 0 1 ## C 0.04 0 0 0 0 ## G 0.00 1 0 0 0 ## T 0.38 0 0 1 0 ## ## [[2]] ## ## Motif name: 2 ## Alternate name: WGATAAG-MEME-1 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 10.2 ## Pseudocount: 1 ## Consensus: WGATAAG ## Target sites: 786 ## E-value: 1.4e-96 ## Extra info: [eval.string] 1.4e-096 ## ## W G A T A A G ## A 0.64 0 1 0 0.96 0.82 0.15 ## C 0.03 0 0 0 0.00 0.00 0.17 ## G 0.01 1 0 0 0.00 0.12 0.63 ## T 0.32 0 0 1 0.04 0.05 0.05 ## ## [[3]] ## ## Motif name: 3 ## Alternate name: AYACH-DREME-2 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 7.73 ## Pseudocount: 1 ## Consensus: ACACH ## Target sites: 1084 ## E-value: 1.5e-34 ## Extra info: [eval.string] 1.5e-034 ## ## A C A C H ## A 1 0.00 1 0 0.38 ## C 0 0.83 0 1 0.34 ## G 0 0.00 0 0 0.00 ## T 0 0.17 0 0 0.28 ## ## [[4]] ## ## Motif name: 4 ## Alternate name: ACAK-DREME-3 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 7.19 ## Pseudocount: 1 ## Consensus: ACAG ## Target sites: 1719 ## E-value: 3.7e-17 ## Extra info: [eval.string] 3.7e-017 ## ## A C A G ## A 1 0 1 0.00 ## C 0 1 0 0.00 ## G 0 0 0 0.76 ## T 0 0 0 0.24 ## ## [[5]] ## ## Motif name: 5 ## Alternate name: CTHATC-DREME-4 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 10.13 ## Pseudocount: 1 ## Consensus: CTHATC ## Target sites: 138 ## E-value: 1.1e-10 ## Extra info: [eval.string] 1.1e-010 ## ## C T H A T C ## A 0 0 0.39 1 0 0 ## C 1 0 0.32 0 0 1 ## G 0 0 0.00 0 0 0 ## T 0 1 0.29 0 1 0 ## ## [[6]] ## ## Motif name: 6 ## Alternate name: YTATCW-MEME-2 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 6.89 ## Pseudocount: 1 ## Consensus: YTATCW ## Target sites: 761 ## E-value: 7.4e-09 ## Extra info: [eval.string] 7.4e-009 ## ## Y T A T C W ## A 0.04 0 0.59 0 0.05 0.37 ## C 0.35 0 0.00 0 0.86 0.05 ## G 0.19 0 0.17 0 0.00 0.01 ## T 0.42 1 0.23 1 0.09 0.57 ## ## [[7]] ## ## Motif name: 7 ## Alternate name: ASWA-DREME-5 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 6.11 ## Pseudocount: 1 ## Consensus: ASWA ## Target sites: 1492 ## E-value: 6e-07 ## Extra info: [eval.string] 6.0e-007 ## ## A S W A ## A 1 0.00 0.7 1 ## C 0 0.42 0.0 0 ## G 0 0.58 0.0 0 ## T 0 0.00 0.3 0 ## ## [[8]] ## ## Motif name: 8 ## Alternate name: ACCCAGW-DREME-6 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 12.13 ## Pseudocount: 1 ## Consensus: ACCCAGW ## Target sites: 51 ## E-value: 0.00036 ## Extra info: [eval.string] 3.6e-004 ## ## A C C C A G W ## A 1 0 0 0 1 0 0.53 ## C 0 1 1 1 0 0 0.00 ## G 0 0 0 0 0 1 0.00 ## T 0 0 0 0 0 0 0.47 ## ## [[9]] ## ## Motif name: 9 ## Alternate name: CMCACCCW-MEME-3 ## Alphabet: DNA ## Type: PPM ## Strands: +- ## Total IC: 12.73 ## Pseudocount: 1 ## Consensus: CMCACCCW ## Target sites: 164 ## E-value: 0.029 ## Extra info: [eval.string] 2.9e-002 ## ## C M C A C C C W ## A 0.01 0.63 0 0.95 0.00 0 0 0.48 ## C 0.96 0.37 1 0.00 0.99 1 1 0.07 ## G 0.00 0.00 0 0.05 0.01 0 0 0.01 ## T 0.03 0.00 0 0.00 0.00 0 0 0.45 ``` --- ## Importing motifs The **universalmotif** package also provides functions to convert between motif objects from different package. Here we convert to TFBStools motif objects. ```r memeMotifsTFBStools <- convert_motifs(memeMotifs, "TFBSTools-PWMatrix") memeMotifsTFBStools ``` ``` ## [[1]] ## An object of class PWMatrix ## ID: HGATA-DREME-1 ## Name: 1 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "7.6e-275" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## W G A T A ## A 1.3294375 -10.272630 2.11689 -10.27263 2.11689 ## C -2.5931079 -10.272630 -10.27263 -10.27263 -10.27263 ## G -10.2726298 1.890255 -10.27263 -10.27263 -10.27263 ## T 0.7069592 -10.272630 -10.27263 2.11689 -10.27263 ## ## [[2]] ## An object of class PWMatrix ## ID: WGATAAG-MEME-1 ## Name: 2 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "1.4e-096" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## W G A T A A G ## A 1.4668977 -9.620220 2.116376 -9.620220 2.060250 1.835703 -0.6292295 ## C -3.1887377 -9.620220 -9.620220 -9.620220 -9.620220 -6.019998 -0.6709083 ## G -4.5166051 1.889768 -9.620220 -9.620220 -9.620220 -1.155168 1.2171203 ## T 0.4818744 -9.620220 -9.620220 2.116376 -2.584504 -2.136796 -2.0684582 ## ## [[3]] ## An object of class PWMatrix ## ID: AYACH-DREME-2 ## Name: 3 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "1.5e-034" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## A C A C H ## A 2.116764 -10.0834793 2.116764 -10.083479 0.7390085 ## C -10.083479 1.6154172 -10.083479 1.890136 0.3243904 ## G -10.083479 -10.0834793 -10.083479 -10.083479 -10.0834793 ## T -10.083479 -0.4093361 -10.083479 -10.083479 0.2690316 ## ## [[4]] ## An object of class PWMatrix ## ID: ACAK-DREME-3 ## Name: 4 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "3.7e-017" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## A C A G ## A 2.117142 -10.748193 2.117142 -10.74819285 ## C -10.748193 1.890495 -10.748193 -10.74819285 ## G -10.748193 -10.748193 -10.748193 1.49966337 ## T -10.748193 -10.748193 -10.748193 0.04283434 ## ## [[5]] ## An object of class PWMatrix ## ID: CTHATC-DREME-4 ## Name: 5 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "1.1e-010" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## C T H A T C ## A -7.118941 -7.118941 0.7598762 2.109777 -7.118941 -7.118941 ## C 1.883507 -7.118941 0.2404110 -7.118941 -7.118941 1.883507 ## G -7.118941 -7.118941 -7.1189411 -7.118941 -7.118941 -7.118941 ## T -7.118941 2.109777 0.3290605 -7.118941 2.109777 -7.118941 ## ## [[6]] ## An object of class PWMatrix ## ID: YTATCW-MEME-2 ## Name: 6 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "7.4e-009" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## Y T A T C W ## A -2.6366821 -9.573647 1.365054357 -9.573647 -2.125646 0.6642764 ## C 0.3796124 -9.573647 -9.573647187 -9.573647 1.671202 -2.3871986 ## G -0.5199509 -9.573647 -0.624335673 -9.573647 -9.573647 -4.3222315 ## T 0.8805593 2.116330 0.005459193 2.116330 -1.384818 1.3064571 ## ## [[7]] ## An object of class PWMatrix ## ID: ASWA-DREME-5 ## Name: 7 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "6.0e-007" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## A S W A ## A 2.117044 -10.5439985 1.5978626 2.117044 ## C -10.543998 0.6423491 -10.5439985 -10.543998 ## G -10.543998 1.1024468 -10.5439985 -10.543998 ## T -10.543998 -10.5439985 0.3915094 -10.543998 ## ## [[8]] ## An object of class PWMatrix ## ID: ACCCAGW-DREME-6 ## Name: 8 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "3.6e-004" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## A C C C A G W ## A 2.096276 -5.70044 -5.70044 -5.70044 2.096276 -5.70044 1.184494 ## C -5.700440 1.87070 1.87070 1.87070 -5.700440 -5.70044 -5.700440 ## G -5.700440 -5.70044 -5.70044 -5.70044 -5.700440 1.87070 -5.700440 ## T -5.700440 -5.70044 -5.70044 -5.70044 -5.700440 -5.70044 1.016094 ## ## [[9]] ## An object of class PWMatrix ## ID: CMCACCCW-MEME-3 ## Name: 9 ## Matrix Class: Unknown ## strand: * ## Pseudocounts: 1 ## Tags: ## $eval.string ## [1] "2.9e-002" ## ## Background: ## A C G T ## 0.2304 0.2696 0.2696 0.2304 ## Matrix: ## C M C A C C C ## A -4.949407 1.4411892 -7.366322 2.029732 -7.366322 -7.366322 -7.366322 ## C 1.831026 0.4618849 1.884707 -7.366322 1.867034 1.884707 1.884707 ## G -7.366322 -7.3663222 -7.366322 -2.262708 -4.292777 -7.366322 -7.366322 ## T -2.861614 -7.3663222 -7.366322 -7.366322 -7.366322 -7.366322 -7.366322 ## W ## A 1.0411226 ## C -1.9808502 ## G -4.2927766 ## T 0.9458359 ``` --- ## Time for an exercise! Exercise on ATACseq data can be found [here](../../exercises/exercises/ATACseq_part3_exercise.html) --- ## Answers to exercise Answers can be found [here](../../exercises/answers/ATACseq_part3_answers.html)