class: center, middle, inverse, title-slide # Introduction to Bioconductor <html> <div style="float:left"> </div> <hr color='#EB811B' size=1px width=796px> </html> ### Rockefeller University, Bioinformatics Resource Centre ### <a href="http://rockefelleruniversity.github.io/Bioconductor_Introduction/" class="uri">http://rockefelleruniversity.github.io/Bioconductor_Introduction/</a> --- # About Bioconductor Bioconductor (BioC) is an open source, community driven software project which provides a framework of tools and databases for the analysis of biological data in R. - Started in 2001 with Robert Gentleman. - Gained popularity through microarray analysis packages (**MArray/Limma**). - Core reviewed and versioned R packages. - Two major releases every year (R has one major release). - Great support for High Throughput sequencing. --- # Bioconductor Goals The broad goals of the Bioconductor project are: - To provide widespread access to a broad range of powerful statistical and graphical methods for the analysis of genomic data. - To facilitate the inclusion of biological metadata in the analysis of genomic data, e.g. literature data from PubMed, annotation data from Entrez genes. - To provide a common software platform that enables the rapid development and deployment of extensible, scalable, and interoperable software. - To further scientific understanding by producing high-quality documentation and reproducible research. - To train researchers on computational and statistical methods for the analysis of genomic data. --- # Bioconductor [website](https://www.bioconductor.org/) <div align="center"> <img src="imgs/bioconductor_2020.png" alt="igv" height="500" width="1000"> </div> --- # Bioconductor packages Bioconductor packages can be broadly split into 4 groups. - Software (Tools for the analysis/visualisation of biological data) - Annotation Data (Tools for the integration of biological metadata) - Experiment Data (Example and actual data from experiments) - Workflow --- # Current Bioconductor Release Current release is Bioconductor 3.13 Includes nearly 2000 software packages. All packages have been - Reviewed. - Tested and evaluated automatically. - Actively maintained and updated. --- # Packages review Before being accepted to Bioconductor all new packages are reviewed so as to pass Bioconductor guidelines. Review includes automatic testing of packages - Testing manual and all examples. - Checking code integrity. --- # Packages review As well as an open review on [Bioconductor github site](https://github.com/Bioconductor/Contributions/issues). 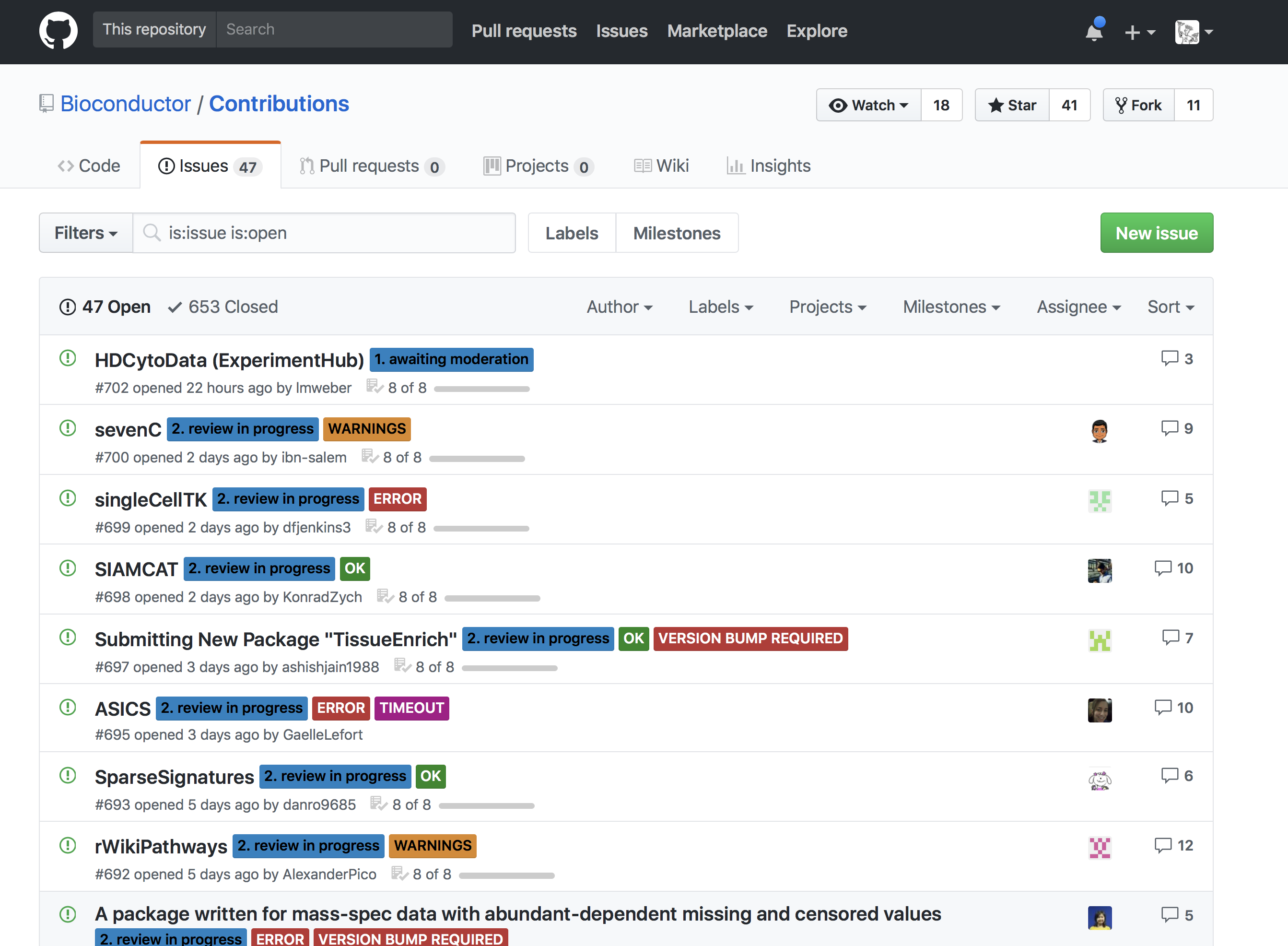 --- # Bioconductor packages Review ensures - All packages can be built in latest version R. - Examples in reference manual can be evaluated. - Vignette manual code can all be run. <div align="center"> <img src="imgs/check_2020.png" alt="igv" height="160" width="600"> </div> --- # Bioconductor packages Example package - [BasecallQC](https://bioconductor.org/packages/release/bioc/html/basecallQC.html) <div align="center"> <img src="imgs/basecallQC_2020.png" alt="igv" height="400" width="400"> </div> --- # Installing Bioconductor and packages Installing Bioconductor/Bioconductor packages is quite straight forward. Every Bioconductor package has a description of the installation R command we can simply copy and paste. Install Bioconductor: ```r if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install(version = "3.10") ``` Install a package: ```r BiocManager::install("basecallQC") ``` --- # Bioconductor package dependencies All dependencies and their required versions are resolved for us. We must be careful however to check the version of Bioconductor we are using. ```r BiocManager::version() ``` ``` ## [1] '3.13' ``` If we wish to update to latest Bioconductor release we can install Bioconductor again: ```r if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install(version = "3.10") ``` --- # Reference Manual .pull-left[ All packages will have a reference manual containing the help pages for every function. This will include importantly - Details of functions' inputs and outputs. - Working examples. ] .pull-right[ <div align="center"> <img src="imgs/manual_2020.png" alt="igv" height="400" width="400"> </div> ] --- # Vignette .pull-left[ All packages will also include at least one vignette. These vignettes detail a typical usage of the package with working examples included. ] .pull-right[ <div align="center"> <img src="imgs/vignette_2020.png" alt="igv" height="400" width="410"> </div> ] --- # Genomics data in Bioconductor Bioconductor packages cover a wide range of biological data types. In this course we are focusing on high throughput sequencing so we will focus on the main packages for this. This includes methods for handling common genomics data types. - Fasta and FastQ - BED, BED6 and narrowPeak/broadPeak - GFF - SAM and BAM --- # FASTA in Bioconductor Genomic sequences stored as [FASTA files](https://rockefelleruniversity.github.io/Genomic_data/gr_course/presentations/slides/GenomicsData.html#9) are handled using the [**Biostrings**](https://bioconductor.org/packages/release/bioc/html/Biostrings.html) package. <div align="center"> <img src="imgs/fasta.png" alt="igv" height="250" width="600"> </div> --- # BED/BED6 in Bioconductor Genomic intervals stored as [BED files](https://rockefelleruniversity.github.io/Genomic_data/r_course/presentations/slides/GenomicsData.html#22) are handled using the [**rtracklayer**](https://bioconductor.org/packages/release/bioc/html/rtracklayer.html) and [**GenomicRanges**](https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html) packages. <div align="center"> <img src="imgs/bed6.png" alt="igv" height="260" width="400"> </div> --- # Wigs and BigWigs in Bioconductor Genomic scores stored as [wig or bigWig files](https://rockefelleruniversity.github.io/Genomic_data/r_course/presentations/slides/GenomicsData.html#25) are handled using the [**rtracklayer**](https://bioconductor.org/packages/release/bioc/html/rtracklayer.html) and [**GenomicRanges**](https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html) packages. <div align="center"> <img src="imgs/wiggle.png" alt="igv" height="500" width="400"> </div> --- # GFF [GFF files](https://rockefelleruniversity.github.io/Genomic_data/r_course/presentations/slides/GenomicsData.html#29) containing gene models are handled using the [**GenomicFeatures**](https://bioconductor.org/packages/release/bioc/html/GenomicFeatures.html) package. <div align="center"> <img src="imgs/gff2.png" alt="igv" height="200" width="600"> </div> --- # FastQ [FastQ files](https://rockefelleruniversity.github.io/Genomic_data/r_course/presentations/slides/GenomicsData.html#12) containing gene models are handled using the [**ShortRead**](https://bioconductor.org/packages/release/bioc/html/ShortRead.html) package. <div align="center"> <img src="imgs/fq1.png" alt="igv" height="200" width="600"> </div> --- # SAM/BAM [SAM and BAM files](https://rockefelleruniversity.github.io/Genomic_data/r_course/presentations/slides/GenomicsData.html#15) are handled using the [**GenomicAlignments**](https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html) package. <div align="center"> <img src="imgs/sam2.png" alt="igv" height="200" width="600"> </div> --- # Reference Data in Bioconductor As well as software packages, we know Bioconductor maintains a number of annotation packages. This includes microarray annotation, gene to ID mappings, genes' functional annotation, genome sequence information and gene/trancript models. --- # Gene annotation Information on model organism's gene annotation is contained with the **org.db** packages. Format is org. **species** . **ID type** .db Homo Sapiens annotation with Entrez Gene IDs -- org.Hs.eg.db <div align="center"> <img src="imgs/orgdb_2020.png" alt="igv" height="400" width="390"> </div> --- # Genome Sequence Genomic sequence information is held within the **BSgenome** packages. Format is BSgenome. **species**. **source**. **major version** Homo Sapiens genome sequence from UCSC's version hg19 -- BSgenome.Hsapiens.UCSC.hg19 <div align="center"> <img src="imgs/bsgenome_2020.png" alt="igv" height="400" width="370"> </div> --- # Gene Models Gene models are held in the **TxDb** packages. Format is TxDb. **species** . **source** . **major version** . **table** Homo Sapiens gene build from UCSC's version hg19 known gene table -- TxDb.Hsapiens.UCSC.hg19.knownGene <div align="center"> <img src="imgs/txdb_2020.png" alt="igv" height="400" width="355"> </div> --- # Time for an exercise. [Link_to_exercises](../../exercises/exercises/biocIntro_exercise.html) [Link_to_answers](../../exercises/answers/biocIntro_answers.html)