class: middle, inverse, title-slide .title[ # Epigenomics, Session 6 ] .subtitle[ ## <html><br /> <br /> <hr color='#EB811B' size=1px width=796px><br /> </html><br /> Bioinformatics Resource Center - Rockefeller University ] .author[ ### <a href="http://rockefelleruniversity.github.io/ATAC.Cut-Run.ChIP/" class="uri">http://rockefelleruniversity.github.io/ATAC.Cut-Run.ChIP/</a> ] .author[ ### <a href="mailto:brc@rockefeller.edu" class="email">brc@rockefeller.edu</a> ] --- ## Recap 1) Fastq QC 2) Alignment 3) Peak calling 4) Technique QC 5) Consensus Building 6) Counting 7) Differentials 8) Annotation and enrichment --- ## This Session - [Motif Databases](https://rockefelleruniversity.github.io/Intro_To_R_1Day/r_course/presentations/singlepage/introToR_Session1.html#motif-databases) - [Visualizing motifs](https://rockefelleruniversity.github.io/Intro_To_R_1Day/r_course/presentations/singlepage/introToR_Session1.html#visualizing-motifs) - [Motif enrichment analysis](https://rockefelleruniversity.github.io/Intro_To_R_1Day/r_course/presentations/singlepage/introToR_Session1.html#motif-enrichment-analysis) - [De novo motif analysis](https://rockefelleruniversity.github.io/Intro_To_R_1Day/r_course/presentations/singlepage/introToR_Session1.html#de-novo-motif-analysis) - [Finding Motifs](https://rockefelleruniversity.github.io/Intro_To_R_1Day/r_course/presentations/singlepage/introToR_Session1.html#finding-motifs) --- ## Our Data We have been working to process and a characterize developmental changes in the context of the TF Sox9 using data from the Fuchs lab: [*The pioneer factor SOX9 competes for epigenetic factors to switch stem cell fates*](https://www.nature.com/articles/s41556-023-01184-y) --- ## Motifs Once we have identified regions of interest from our ATAC or Cut&Run often the next step is to investigate the motifs enriched under peaks. Motif analysis like this can help find the drivers of epigenomic changes and help create a more mechanistic understanding of your experiment. For Cut&Run with a known transcription factor this kind of analysis may be less obvious as we have an expected target i.e. Sox9. That said it is still useful to validate our IP, find cofactors, indirect effects and also find specific motif variants. --- class: inverse, center, middle # Motif Databases <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Known Motif sources Bioconductor provides two major sources of motifs as database packages. These include: * The [MotifDb](https://www.bioconductor.org/packages/release/bioc/html/MotifDb.html) package. * The JASPAR databases, [JASPAR2024](https://www.bioconductor.org/packages/release/data/annotation/html/JASPAR2024.html) being latest (they do biannual updates). --- ## MotifDb The MotifDB package collects motif information from a wide range of sources and stores them in a DB object for use with other Bioconductor packages. ``` r library(MotifDb) ``` ``` ## Warning: package 'MotifDb' was built under R version 4.4.1 ``` ``` ## Warning in load(data.file): strings not representable in native encoding will ## be translated to UTF-8 ``` ``` ## See system.file("LICENSE", package="MotifDb") for use restrictions. ``` ``` r MotifDb ``` ``` ## MotifDb object of length 12657 ## | Created from downloaded public sources, last update: 2022-Mar-04 ## | 12657 position frequency matrices from 22 sources: ## | FlyFactorSurvey: 614 ## | HOCOMOCOv10: 1066 ## | HOCOMOCOv11-core-A: 181 ## | HOCOMOCOv11-core-B: 84 ## | HOCOMOCOv11-core-C: 135 ## | HOCOMOCOv11-secondary-A: 46 ## | HOCOMOCOv11-secondary-B: 19 ## | HOCOMOCOv11-secondary-C: 13 ## | HOCOMOCOv11-secondary-D: 290 ## | HOMER: 332 ## | JASPAR_2014: 592 ## | JASPAR_CORE: 459 ## | ScerTF: 196 ## | SwissRegulon: 684 ## | UniPROBE: 380 ## | cisbp_1.02: 874 ## | hPDI: 437 ## | jaspar2016: 1209 ## | jaspar2018: 1564 ## | jaspar2022: 1956 ## | jolma2013: 843 ## | stamlab: 683 ## | 62 organism/s ## | Hsapiens: 6075 ## | Mmusculus: 1554 ## | Dmelanogaster: 1437 ## | Athaliana: 1371 ## | Scerevisiae: 1221 ## | NA: 184 ## | other: 815 ## Scerevisiae-ScerTF-ABF2-badis ## Scerevisiae-ScerTF-CAT8-badis ## Scerevisiae-ScerTF-CST6-badis ## Scerevisiae-ScerTF-ECM23-badis ## Scerevisiae-ScerTF-EDS1-badis ## ... ## Mmusculus-UniPROBE-Zfp740.UP00022 ## Mmusculus-UniPROBE-Zic1.UP00102 ## Mmusculus-UniPROBE-Zic2.UP00057 ## Mmusculus-UniPROBE-Zic3.UP00006 ## Mmusculus-UniPROBE-Zscan4.UP00026 ``` --- ## MotifDb MotifDb object is special class of object called a **MotifList**. ``` r class(MotifDb) ``` ``` ## [1] "MotifList" ## attr(,"package") ## [1] "MotifDb" ``` --- ## MotifDb Like standard List objects we can use length and names to get some information on our object ``` r length(MotifDb) ``` ``` ## [1] 12657 ``` ``` r MotifNames <- names(MotifDb) MotifNames[1:10] ``` ``` ## [1] "Scerevisiae-ScerTF-ABF2-badis" "Scerevisiae-ScerTF-CAT8-badis" ## [3] "Scerevisiae-ScerTF-CST6-badis" "Scerevisiae-ScerTF-ECM23-badis" ## [5] "Scerevisiae-ScerTF-EDS1-badis" "Scerevisiae-ScerTF-FKH2-badis" ## [7] "Scerevisiae-ScerTF-FZF1-badis" "Scerevisiae-ScerTF-GIS1-badis" ## [9] "Scerevisiae-ScerTF-GSM1-badis" "Scerevisiae-ScerTF-GZF3-badis" ``` --- ## Accesing MotifDb contents We can also access information directly from our list using standard list accessors. Here a **[** will subset to a single MotifList. Now we can see the information held in the MotifList a little more clearly. ``` r MotifDb[1] ``` ``` ## MotifDb object of length 1 ## | Created from downloaded public sources, last update: 2022-Mar-04 ## | 1 position frequency matrices from 1 source: ## | ScerTF: 1 ## | 1 organism/s ## | Scerevisiae: 1 ## Scerevisiae-ScerTF-ABF2-badis ``` --- ## Accesing MotifDb contents A **[[** will subset to object within the element as with standard lists. Here we extract the position probability matrix. ``` r MotifDb[[1]] ``` ``` ## 1 2 3 4 5 6 ## A 0.09 0.01 0.01 0.97 0.01 0.94 ## C 0.09 0.97 0.01 0.01 0.01 0.02 ## G 0.02 0.01 0.01 0.01 0.97 0.02 ## T 0.80 0.01 0.97 0.01 0.01 0.02 ``` ``` r colSums(MotifDb[[1]]) ``` ``` ## 1 2 3 4 5 6 ## 1 1 1 1 1 1 ``` --- ## Accesing MotifDb contents We can extract a DataFrame of all the motif metadata information using the **values()** function. ``` r values(MotifDb)[1:2, ] ``` ``` ## DataFrame with 2 rows and 15 columns ## providerName providerId dataSource geneSymbol ## <character> <character> <character> <character> ## Scerevisiae-ScerTF-ABF2-badis badis.ABF2 ABF2 ScerTF ABF2 ## Scerevisiae-ScerTF-CAT8-badis badis.CAT8 CAT8 ScerTF CAT8 ## geneId geneIdType proteinId proteinIdType ## <character> <character> <character> <character> ## Scerevisiae-ScerTF-ABF2-badis YMR072W SGD Q02486 UNIPROT ## Scerevisiae-ScerTF-CAT8-badis YMR280C SGD P39113 UNIPROT ## organism sequenceCount bindingSequence ## <character> <character> <character> ## Scerevisiae-ScerTF-ABF2-badis Scerevisiae NA NA ## Scerevisiae-ScerTF-CAT8-badis Scerevisiae NA NA ## bindingDomain tfFamily experimentType ## <character> <character> <character> ## Scerevisiae-ScerTF-ABF2-badis NA NA NA ## Scerevisiae-ScerTF-CAT8-badis NA NA NA ## pubmedID ## <character> ## Scerevisiae-ScerTF-ABF2-badis 19111667 ## Scerevisiae-ScerTF-CAT8-badis 19111667 ``` --- ## Accesing MotifDb contents We can use the **query** function to subset our MotifList by infomation in the metadata. ``` r Sox9Motifs <- query(MotifDb, "Sox9") ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6084 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6140 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6281 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6598 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6622 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` r Sox9Motifs ``` ``` ## MotifDb object of length 18 ## | Created from downloaded public sources, last update: 2022-Mar-04 ## | 18 position frequency matrices from 11 sources: ## | HOCOMOCOv10: 2 ## | HOCOMOCOv11-core-B: 1 ## | HOCOMOCOv11-secondary-B: 1 ## | HOMER: 1 ## | JASPAR_2014: 1 ## | JASPAR_CORE: 1 ## | SwissRegulon: 1 ## | jaspar2016: 1 ## | jaspar2018: 1 ## | jaspar2022: 1 ## | jolma2013: 7 ## | 3 organism/s ## | Hsapiens: 16 ## | Mmusculus: 1 ## | other: 1 ## Hsapiens-SwissRegulon-SOX9.SwissRegulon ## Hsapiens-HOCOMOCOv10-SOX9_HUMAN.H10MO.B ## Mmusculus-HOCOMOCOv10-SOX9_MOUSE.H10MO.B ## Hsapiens-HOCOMOCOv11-core-B-SOX9_HUMAN.H11MO.0.B ## Hsapiens-HOCOMOCOv11-secondary-B-SOX9_HUMAN.H11MO.1.B ## ... ## Hsapiens-jolma2013-SOX9-3 ## Hsapiens-jolma2013-SOX9-4 ## Hsapiens-jolma2013-SOX9-5 ## Hsapiens-jolma2013-SOX9-6 ## Hsapiens-jolma2013-SOX9-7 ``` --- ## Accesing MotifDb contents For more specific queries, multiple words can be used for filtering. ``` r Sox9Motifs <- query(MotifDb, c("Sox9", "hsapiens", "jaspar2022")) ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6084 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6140 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6281 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6598 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6622 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6084 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6140 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6281 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6598 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6622 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6084 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6140 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6281 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6598 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6622 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` r Sox9Motifs ``` ``` ## MotifDb object of length 1 ## | Created from downloaded public sources, last update: 2022-Mar-04 ## | 1 position frequency matrices from 1 source: ## | jaspar2022: 1 ## | 1 organism/s ## | Hsapiens: 1 ## Hsapiens-jaspar2022-SOX9-MA0077.1 ``` --- ## JASPAR2020 package The JASPAR packages are updated more frequently and so may contain motifs not characterized in the MotifDb package. We can load the **JASPAR** package as usual. This is a little more complex then MotifDb, but ultimately we are connecting to a SQL database. ``` r library(JASPAR2024) ``` ``` ## Loading required package: BiocFileCache ``` ``` ## Warning: package 'BiocFileCache' was built under R version 4.4.1 ``` ``` ## Loading required package: dbplyr ``` ``` r library(RSQLite) JASPAR2024 <- JASPAR2024() ``` ``` ## adding rname 'https://testjaspar.uio.no/download/database/JASPAR2024.sqlite' ``` ``` r sq24 <- RSQLite::dbConnect(RSQLite::SQLite(), db(JASPAR2024)) ``` --- ## JASPAR2020 and TFBSTools To interact with JASPAR package we will make use of the **TFBSTools** package from the same lab. Whereas the JASPAR package holds the information on Motifs and Position Probability Matrices (PPMs), TFBSTools has the functionality to manipulate and interact with these tools. Three useful functions available from TFBStools to interact with the JASPAR databases are the **getMatrixSet**, **getMatrixByID** and **getMatrixByID**. ``` r library(TFBSTools) `?`(getMatrixByID) ``` --- ## TFBSTools for TFs The **getMatrixByID** and **getMatrixByName** take the JASPAR DB object and a JASPAR ID or transcription factor name respectively. If you are unsure of exact names/IDs you can review the Jaspar [website](https://jaspar.elixir.no/matrix/MA0077.1/) first. Here we are using the transcription factor SOX9 again. The result is a new object class **PFMatrix**. ``` r SOX9mat <- getMatrixByName(sq24, "SOX9") class(SOX9mat) ``` ``` ## [1] "PFMatrix" ## attr(,"package") ## [1] "TFBSTools" ``` --- ## TFBSTools for motifs JASPAR IDs are unique, so we can use them to select the exact motif we wanted. Sox9 has two very subtlely different motifs ``` r SOX9mat2 <- getMatrixByID(sq24, "MA0077.1") ``` --- ## TFBSTools for motifs List accessors will not work here but we can retrieve names using the **ID** function. ``` r ID(SOX9mat) ``` ``` ## [1] "MA0077.2" ``` --- ## Position Frequency Matrix To get hold of the Position Frequency Matrix (PFM) we can use the **Matrix** or **as.matrix** functions. ``` r myMatrix <- Matrix(SOX9mat) myMatrixToo <- as.matrix(myMatrix) myMatrix ``` ``` ## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] ## A 3 8 71 2 7 2 13 9 ## C 55 38 1 1 0 2 0 7 ## G 9 6 0 2 4 72 4 6 ## T 9 24 4 71 65 0 59 54 ``` --- ## TFBSTools for motif sets We can also use the **getMatrixSet()** function to retrieve sets of motifs. We can specify a list of options for motifs we want to retrieve. To see the available filters use the help for getMatrixSet() function, ?getMatrixSet. Here we retrieve the vertebrate, JASPAR CORE motifs. We additional specify *all_versions* is FALSE to only include the latest version of a motif. ``` r opts <- list() opts[["collection"]] <- "CORE" opts[["tax_group"]] <- "vertebrates" opts[["all_versions"]] <- FALSE motifList <- getMatrixSet(sq24, opts) ``` --- class: inverse, center, middle # Visualizing motifs <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Visualizing motifs The seqLogo package offers a simple and intuitive way of visualizing our base frequencies within out motifs using the **seqLogo** function. A simple seqLogo shows the relative frequency of a base at each motif position by the relative size of the base compared to other bases at the same position. <!-- --> --- ## Visualizing motifs To create a seqLogo we must supply a PPM to the **seqLogo** function. Here we grab a PPM straight from MotifdDb for Sox9. Here we set the **ic.scale** to FALSE to show the probability across the Y-axis ``` r library(seqLogo) Sox9Motifs <- query(MotifDb, "Sox9") ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6084 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6140 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6281 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6598 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): input string 6622 is invalid ``` ``` ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ## Warning in grep(queryString, mcols(object)[, colname], ignore.case = ## ignore.case): unable to translate 'Three-zinc finger Kr<c3><bc>ppel-related ## factors' to a wide string ``` ``` r seqLogo::seqLogo(Sox9Motifs[[1]], ic.scale = FALSE) ``` <!-- --> --- ## Visualizing motifs It may be useful to plot the probabilities as information content. Information content here will range from 0 to 2 Bits. Positions with equal probabilities for each base will score 0 and positions with only 1 possible base will score 2. This allows you to quickly identify the important bases. ``` r seqLogo::seqLogo(Sox9Motifs[[1]]) ``` 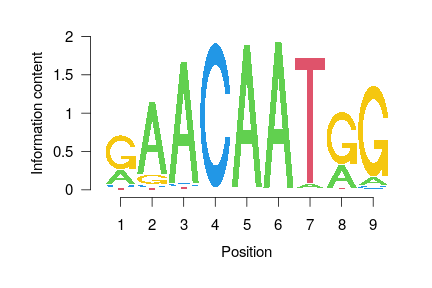<!-- --> --- ## Visualizing motifs The TFBSTools package and JASPAR provide us with point frequency matrices which we can't use directly in the seqLogo package ``` r myMatrix ``` ``` ## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] ## A 3 8 71 2 7 2 13 9 ## C 55 38 1 1 0 2 0 7 ## G 9 6 0 2 4 72 4 6 ## T 9 24 4 71 65 0 59 54 ``` --- ## Visualizing motifs We can convert our point frequency matrix to a point probabilty matrix by simply dividing columns by their sum. ``` r ppm <- myMatrix/colSums(myMatrix) ppm ``` ``` ## [,1] [,2] [,3] [,4] [,5] [,6] [,7] ## A 0.03947368 0.10526316 0.93421053 0.02631579 0.09210526 0.02631579 0.17105263 ## C 0.72368421 0.50000000 0.01315789 0.01315789 0.00000000 0.02631579 0.00000000 ## G 0.11842105 0.07894737 0.00000000 0.02631579 0.05263158 0.94736842 0.05263158 ## T 0.11842105 0.31578947 0.05263158 0.93421053 0.85526316 0.00000000 0.77631579 ## [,8] ## A 0.11842105 ## C 0.09210526 ## G 0.07894737 ## T 0.71052632 ``` --- ## Visualizing motifs We can then plot the result matrix using seqLogo. ``` r seqLogo::seqLogo(ppm) ``` <!-- --> --- ## Visualizing motifs Fortunately, TFBSTools has its own version of seqLogo function we can use with one of its own **ICMatrix** classes. We simply need to convert our object with the **toICM()** function. ``` r Sox9_IC <- toICM(SOX9mat) TFBSTools::seqLogo(Sox9_IC) ``` <!-- --> --- ## Visualizing with ggplot We can also use a ggplot2 style grammar of graphics with the **ggseqlogo** package. ``` r library(ggseqlogo) library(ggplot2) ggseqlogo(myMatrix) + theme_minimal() ``` ``` ## Warning: The `<scale>` argument of `guides()` cannot be `FALSE`. Use "none" instead as ## of ggplot2 3.3.4. ## i The deprecated feature was likely used in the ggseqlogo package. ## Please report the issue at <https://github.com/omarwagih/ggseqlogo/issues>. ## This warning is displayed once every 8 hours. ## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was ## generated. ``` <!-- --> --- class: inverse, center, middle # Motif enrichment analysis <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Motif review Motif enrichment can be performed using the [MEME suite](https://meme-suite.org/meme/) of tools. They have developed a huge collection of algorithms with situation specificity for finding and matching motifs or detecting differential enrichment of motifs. You can directly run these tools sing the web interface (and we show you how to work with the results [here](https://rockefelleruniversity.github.io/RU_ATACseq/presentations/singlepage/RU_ATAC_part3.html)), but in these sessions we will show you how to run this through R. --- ## Installing MEME As we did for MACS, we can also install MEME from the Conda repository using the Herper package. ``` r library(Herper) install_CondaTools("meme", "motif_analysis", channels = "bioconda", pathToMiniConda = "miniconda") ``` --- ## Motif DBs for MEME Now we have MEME installed, the next step is to grab some Motifs as our reference. We can follow similar steps to before. ``` r opts <- list() opts[["tax_group"]] <- "vertebrates" opts[["collection"]] <- "CORE" opts[["all_versions"]] <- FALSE jaspar <- JASPAR2024() sq24 <- RSQLite::dbConnect(RSQLite::SQLite(), db(jaspar)) ``` --- ## Motif DBs for MEME We specifically will want to convert this to a new format: the *universalmotif* class. We can do this by grabbing our motifs like we did for TFBStools, then using the *convert_motifs* function. The result is a list of universalmotif PCMs. ``` r library(universalmotif) ``` ``` ## Warning: package 'universalmotif' was built under R version 4.4.1 ``` ``` r motifs <- getMatrixSet(sq24, opts) motifs_um <- convert_motifs(motifs) class(motifs_um) ``` ``` ## [1] "list" ``` ``` r class(motifs_um[[1]]) ``` ``` ## [1] "universalmotif" ## attr(,"package") ## [1] "universalmotif" ``` --- ## Query Sequences for MEME We need a collection of sequences that we are querying. In this case our query is: what motifs are enriched in our regulated regions at W6 time point. We can start by importing the ranges from the data folder *UpinW6.bed*. ``` r library(rtracklayer) UpinW6_peaks <- rtracklayer::import("data/UpinW6.bed") ``` --- ## Query Sequences for MEME Next we need to grab the corresponding sequence from under these ranges. Where you are grabbing sequences from needs to match your alignment reference. If you have updated your BSGenome or are using a different build you can grab the sequence ifnromation from any fasta using *readDNAStringSet* ``` r library(Biostrings) library(BSgenome.Mmusculus.UCSC.mm10) peaksSequences <- getSeq(BSgenome.Mmusculus.UCSC.mm10, UpinW6_peaks) names(peaksSequences) <- paste0("peak_", 1:length(UpinW6_peaks)) ``` --- ## Background Sequences for MEME The last thing we need is background sequences. Though you can use a random shuffled background it is often better to give something a little more representative of your data i.e. consensus peaks. Otherwise common motifs found in regions like promoters may seem enriched, even if they are not enriched in the specific promoters you care about. ``` r HC_peaks <- rtracklayer::import("data/HC_Peaks.bed") background_peaksSequences <- getSeq(BSgenome.Mmusculus.UCSC.mm10, HC_peaks) names(background_peaksSequences) <- paste0("peak_", 1:length(HC_peaks)) ``` --- ## Running MEME Though there is no R pacakge for MEME, there is a wrapper package that will run MEME for you from within R. This is called *memes*. We just need to be able to tell memes where MEME is. ``` r library(memes) meme_path_id = "miniconda/envs/motif_analysis/bin" ``` --- ## Running MEME We will use the [**Ame** algorithm](https://meme-suite.org/meme/tools/ame) (Analysis of Motif Enrichment) to look for enriched motifs. *NOTE: This will take some time. We will provide the result* ``` r ame_results <- runAme(peaksSequences, control = background_peaksSequences, outdir = "ame_diff", meme_path = meme_path_id, database = motifs_um) ``` --- ## Reviewing AME results Once this has run the MEME results are saved in the output directory we specified in the function. *data/ame_diff*. We can review the [report]("data/ame_diff/ame.html") to see our top hits. --- ## Reviewing AME results The memes package also returns our results into R. We have saved the R result object *data/ame_results.RData* so you can load it in. This object is a special kind of data frame called a tibble. This means we can use a combination of tidyverse and memes functions to extract our result and generate some nice plots. ``` r load(file = "data/ame_results.RData") class(ame_results) ``` ``` ## [1] "spec_tbl_df" "tbl_df" "tbl" "data.frame" ``` --- ## Reviewing AME results First lets look at our top 10 motifs associated with the W6 timepoint. They are predominately the SOX family, which is a good result. ``` r ame_results %>% dplyr::filter(rank %in% 1:10) %>% plot_ame_heatmap() + ggtitle("Top 10 AME Hits") ``` <!-- --> --- ## Reviewing AME results We can then use what we know about making motifs to quickly grab our seqlogo. ``` r top_res_motif <- getMatrixByID(sq24, ame_results$motif_alt_id[1]) top_res_motif_mat <- Matrix(top_res_motif) ggseqlogo(top_res_motif_mat) + theme_minimal() ``` <!-- --> --- ## A quick note As you can see we have a high degree of redundancy in our results i.e. all the top hits are different Sox! This can make it a little trickier to aprse your results. Two factors can help with this. 1) Filter out unexpressed genes. It is possible to use matching RNAseq (or even ATAC) to filter out TF motifs if they are not expressed. This will also boost your padj scores. 2) There are [JASPAR familial profiles](https://jaspar.elixir.no/matrix-clusters/) that contain a consensus motif for TF families. This will simplify the result, though it may blunt and obfuscate some findings. --- class: inverse, center, middle # De novo motif analysis <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## De novo motifs Databases of motifs are a great resource, but sometimes they fall short or miss aditional biology for various reasons. * Uncharacterized motif * Rare motif variants * Motif evolution versus model organism * Cofactor binding By doing de novo motif characterization you can account for this. The [*DREME* algorithm](https://meme-suite.org/meme/tools/dreme) from MEME is used for this. --- ## Running DREME We can run DREME (Discriminative Regular Expression Motif Elicitation) in a very similar way to AME, using the *runDreme()* function. The only change is we do not include a motif reference database. ``` r dreme_results <- runDreme(peaksSequences, control = background_peaksSequences, outdir = "dreme_diff", meme_path = meme_path_id) ``` --- ## Reviewing Dreme results As before, once this has run the MEME results are saved in the output directory we specified in the function. *data/dreme_diff*. We can review the [report]("data/dreme_diff/dreme.html") to see our top hits. --- ## Reviewing DREME results The memes package also returns our results into R. We have saved the R result object *data/dreme_results.RData* so you can load it in. This object again is a special kind of data frame, but this time it is a universalmotif data frame as it has motifs stored inside. ``` r load(file = "data/dreme_results.RData") class(dreme_results) ``` ``` ## [1] "universalmotif_df" "data.frame" ``` --- ## Reviewing DREME results DREME results will be sightly different to AME as we do not know the identity of the motif. But we can view the motifs easily using some universalmotif functions. ``` r dreme_results[1:3, ] %>% to_list() %>% view_motifs() ``` <!-- --> --- ## Matching DREME motifs Once you have derived a list of de novo motifs, the next step is then to compare them to a list of known motifs. We can use the [*TomTom*](https://meme-suite.org/meme/tools/tomtom) algorithm from MEME to do this. ``` r tomtom_results <- runTomTom(dreme_results, meme_path = meme_path_id, database = motifs_um, outdir = "tomtom_from_dreme") ``` --- ## Reviewing TOMTOM results The *best_match_motif* column contains all the top hits from TOMTOM for each de novo motif. ``` r load(file = "data/tomtom_results.RData") view_motifs(tomtom_results$best_match_motif[1:3]) ``` <!-- --> --- ## Reviewing TOMTOM results Though there is a top hit, each motif can have multiple hits. These are some of the many potential hits: ``` r unlist(lapply(tomtom_results$tomtom[[1]]$match_motif, function(x) x@name)) ``` ``` ## [1] "SOX4" "Sox6" "SOX10" "SOX13" "SOX15" ## [6] "Sox3" "SOX2" "Sox11" "Sox5" "Sox7" ## [11] "Sox17" "Dmrt1" "Sox1" "SOX8" "POU2F1::SOX2" ## [16] "SOX18" "SOX14" "SOX21" "SOX9" "SRY" ## [21] "Hnf1A" "Lef1" "TCF7L1" "TCF7" ``` --- ## Reviewing TOMTOM results If you wanted to review these motifs you can use the *view_tomtom_hits* function. This allows you to compare the motifs easily to the original. Though our result is pretty clear, this is an important step as the "best match" is not always the right match. You need to make sure it amkes sense given your domain knowledge. ``` r tomtom_results[1, ] %>% view_tomtom_hits(top_n = 3) ``` --- class: inverse, center, middle # Finding Motifs <html><div style='float:left'></div><hr color='#EB811B' size=1px width=720px></html> --- ## Motif Locations We now have a couple of tools in our toolkit to characterize which motifs are interesting to us. Often the next step is to actually determine where those motifs are. Unsurprisingly, MEME also has a tool for this: [FIMO](https://meme-suite.org/meme/tools/fimo). You can use the [*runFIMO()*](https://www.bioconductor.org/packages/devel/bioc/vignettes/memes/inst/doc/core_fimo.html) function to do this through memes. There is also a nice alternative tool called motifmatchr, which we will show you as it is very quick. --- ## motifmatchr To identify known motifs we will use the **motifmatchr** package which is a wrapper for the MOODS c++ library. This means that motifmatchr offers us a fast methods to identify motifs within our data. We will use motifmatchr/MOODs with a default p-value cut-off. --- ## motifmatchr Again lets use the JASPAR database as our reference for this. For motifmatchr we want a PFMatrixList using TFBSTools. ``` r opts <- list() opts[["tax_group"]] <- "vertebrates" opts[["collection"]] <- "CORE" opts[["all_versions"]] <- FALSE jaspar <- JASPAR2024() sq24 <- RSQLite::dbConnect(RSQLite::SQLite(), db(jaspar)) motifs <- getMatrixSet(sq24, opts) ``` --- ## motifmatchr The motifmatchr package main function is **matchMotifs()**. As with many Bioconductor functions , **matchMotifs** makes use of other Bioconductor objects such the BSGenome, GRanges and summarizedExperiment objects. We can review the full functionality by reading the help for matchMotifs, ?matchMotifs. --- ## motifmatchr inputs motifmatchr needs some search space. Though we could run this genome-wide, for efficiency and speeds sake we normally just run it on regions of interest i.e. our W6-specific peaks. Specifically we need the sequence associated with these peaks. Luckily we generated all this while running MEME. ``` r peaksSequences ``` ``` ## DNAStringSet object of length 4652: ## width seq names ## [1] 513 AAGGCCTGTCAAAAATCTTAAC...GGAGTTTGAGCCTTGAAAGCT peak_1 ## [2] 1017 ATCCCCATGAATTTCACTACCT...GCTTGTGAGTAGAGATCTCCT peak_2 ## [3] 448 TGAGGAGTCAATGTTTTTTAGC...CTATGCCTTTAACCCATGGAT peak_3 ## [4] 744 AGACAAAATGAGCCCTAGTGGT...CCTGATAAACACTGCCTGTCT peak_4 ## [5] 573 TGCCTCCTCTAAACTCCTCTGC...TCCAAAGGCATCTTCAGCTGT peak_5 ## ... ... ... ## [4648] 595 AGAAACACAGTCGGACACAGGG...ACGGAGATGATTCACATCCCT peak_4648 ## [4649] 350 ATGCAAACATACCTTCCCCCTA...AAAACAAAACACCCAAAACAT peak_4649 ## [4650] 403 AGCAGCTGGAGAGAGAGAGATG...AAGCAATCATTCGTCTTCTCT peak_4650 ## [4651] 383 AGGTTTGTGTGCAGGAAGAACT...AGCCATCGTGACTGTTGCCCA peak_4651 ## [4652] 171 TAAGGTAGTCATTTTCTGGGAT...TGGTATGCATCCTAGTACACT peak_4652 ``` --- ## Finding motif positions As we saw in its help, The matchMotifs function can provide output of motif matches as matches, scores or positions. Here we scan for the top 3 motifs from our AME results using the sequence under the first 100 peaks and specify the out as **positions**. ``` r library(motifmatchr) ``` ``` ## Warning: package 'motifmatchr' was built under R version 4.4.1 ``` ``` r motifs_subset <- motifs[names(motifs) %in% ame_results$motif_alt_id[1:3]] motif_positions <- matchMotifs(motifs_subset, peaksSequences[1:100], out = "positions") class(motif_positions) ``` ``` ## [1] "list" ``` ``` r length(motif_positions) ``` ``` ## [1] 3 ``` --- ## Finding motif positions The result contains a list of the same length as the number of motifs tested. Each element contains a IRangeslist with an entry for every sequence tested and a IRanges of motif positions within the peak sequences. ``` r motif_positions$MA1152.2 ``` ``` ## IRangesList object of length 100: ## [[1]] ## IRanges object with 0 ranges and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## ## [[2]] ## IRanges object with 0 ranges and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## ## [[3]] ## IRanges object with 1 range and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## [1] 295 301 7 | + 11.582 ## ## ... ## <97 more elements> ``` --- ## Finding motif positions We can unlist our IRangeslist to a standard list for easier working. ``` r MA1152.2hits <- motif_positions$MA1152.2 names(MA1152.2hits) <- names(peaksSequences[1:100]) unlist(MA1152.2hits, use.names = TRUE) ``` ``` ## IRanges object with 24 ranges and 2 metadata columns: ## start end width | strand score ## <integer> <integer> <integer> | <character> <numeric> ## peak_3 295 301 7 | + 11.582 ## peak_7 87 93 7 | + 11.582 ## peak_9 654 660 7 | - 11.582 ## peak_14 313 319 7 | + 11.582 ## peak_19 320 326 7 | + 11.582 ## ... ... ... ... . ... ... ## peak_79 132 138 7 | + 11.582 ## peak_89 164 170 7 | + 11.582 ## peak_94 111 117 7 | - 11.582 ## peak_94 186 192 7 | - 11.582 ## peak_96 79 85 7 | + 11.582 ``` --- ## Using motif positions Once motif instances have been identified they can then be used to generate metaplots like we saw in [session 2 with soGGi](https://rockefelleruniversity.github.io/ATAC.Cut-Run.ChIP/presentations/singlepage/Session3_QC.html#Evaluating_TSS_signal), or we could use [profileplyr](https://www.bioconductor.org/packages/devel/bioc/vignettes/profileplyr/inst/doc/profileplyr.html#generate-group-annotated-heatmap-in-r-directly-with-generateenrichedheatmap). These metaplots would show changes to accessibility/enrichment directly over the motifs. The only changes to what we should in session 2 would be to replace the TSS ranges with motif ranges. --- ## Finding motif hits We may simply want to map motifs to their peaks. To do this we can set the **out** parameter to **matches**. This will return a SummarizedExperiment object. ``` r motifHits <- matchMotifs(motifs, peaksSequences, out = "matches") class(motifHits) ``` ``` ## [1] "SummarizedExperiment" ## attr(,"package") ## [1] "SummarizedExperiment" ``` ``` r motifHits ``` ``` ## class: SummarizedExperiment ## dim: 4652 879 ## metadata(0): ## assays(1): motifMatches ## rownames: NULL ## rowData names(0): ## colnames(879): MA0004.1 MA0069.1 ... MA1602.2 MA1722.2 ## colData names(1): name ``` --- ## Finding motif hits We can retrieve a matrix of matches by motif and peak using the motifMatches function. ``` r mmMatrix <- motifMatches(motifHits) dim(mmMatrix) ``` ``` ## [1] 4652 879 ``` ``` r mmMatrix[1:8, 1:8] ``` ``` ## 8 x 8 sparse Matrix of class "lgCMatrix" ## MA0004.1 MA0069.1 MA0071.1 MA0074.1 MA0101.1 MA0107.1 MA0111.1 MA0115.1 ## [1,] . . . . . . . . ## [2,] . . | . . . | | ## [3,] . . . . . . . . ## [4,] . . | . . . . | ## [5,] | . . . | | . . ## [6,] . . . . . . . . ## [7,] . . . . . . | . ## [8,] . . . . | . . . ``` --- ## Finding motif hits Although a sparse matrix, we can still use our matrix operations to extract useful information from this object. We can use the **colSums()** to identify the total occurrence of motifs in our peak sequences. Fittingly our top hit from AME has a high number of motifs. ``` r totalMotifOccurence <- colSums(mmMatrix) totalMotifOccurence[1:4] totalMotifOccurence["MA1152.2"] ``` ``` ## MA0004.1 MA0069.1 MA0071.1 MA0074.1 ## 242 106 183 147 ``` ``` ## MA1152.2 ## 674 ``` --- ## Finding motif hits We can also identify peaks which contain a hit for a selected motif. ``` r UpinW6_peaksWithMA1152.2 <- UpinW6_peaks[mmMatrix[, "MA1152.2"] == 1] UpinW6_peaksWithMA1152.2 ``` ``` ## GRanges object with 674 ranges and 2 metadata columns: ## seqnames ranges strand | name score ## <Rle> <IRanges> <Rle> | <character> <numeric> ## [1] chr7 114358259-114358706 * | <NA> 0 ## [2] chr6 52684371-52684890 * | <NA> 0 ## [3] chr6 134329482-134330318 * | <NA> 0 ## [4] chr14 123530967-123531638 * | <NA> 0 ## [5] chr10 24407882-24408394 * | <NA> 0 ## ... ... ... ... . ... ... ## [670] chr14 70211676-70212181 * | <NA> 0 ## [671] chr4 124400836-124401853 * | <NA> 0 ## [672] chr14 78531771-78532053 * | <NA> 0 ## [673] chr4 100174849-100175341 * | <NA> 0 ## [674] chr15 91037086-91037371 * | <NA> 0 ## ------- ## seqinfo: 20 sequences from an unspecified genome; no seqlengths ``` --- ## Other motif tools We have focussed primarily on using MEME to conduct our analysis, there are several other popular tools. * [*Homer*](http://homer.ucsd.edu/homer/motif/) has some motif detection tools that are conceptually similar to MEME. * [*chromVAR*](https://github.com/GreenleafLab/chromVAR) continues on from what we have done with motifmatchr and checks for differential accessibility of peaks that do/don't contain motifs. This package is designed for ATAC and we have a guide on rnning this [here](https://rockefelleruniversity.github.io/RU_ATACseq/presentations/singlepage/RU_ATAC_part3.html#summarizing_ATAC_signal_to_Motifs) --- ## Time for an exercise! Exercise on functions can be found [here](https://rockefelleruniversity.github.io/ATAC.Cut-Run.ChIP/exercises/exercises/MyExercise6_exercise.html) --- ## Answers to exercise. Answers can be found here [here](https://rockefelleruniversity.github.io/ATAC.Cut-Run.ChIP/exercises/answers/MyExercise6_answers.html)